
Abstract
Despite extensive efforts to identify a magic bullet to prevent the occurrence or mitigate the progression of Alzheimer’s disease (AD), no effective single “pill-for-an-ill” strategy has been found, which is likely why on April 28, 2010, an NIH consensus panel on AD prevention pronounced, “There is no good evidence that Alzheimer’s disease or the other forms of dementia affecting millions of Americans are preventable…there are no modifiable issues or variables that are going to prevent Alzheimer’s or cognitive decline, and people should know that.”
We beg to differ. A clear association has been well documented in the peer-reviewed medical literature between lifestyle habits—diet, intake of specific micronutrients, and exercise–and the occurrence of AD. This review summarizes key, clinically relevant, findings.
Statins—Not the Answer to Alzheimer’s Disease
As noted in Part I of this review, as our understanding of the etiology of AD has deepened, the evidence has become incontrovertible that no single molecular target or pharmaceutical silver bullet will prevent or cure AD. Nevertheless, it has been suggested that, via their cholesterol-lowering effects, statins might lessen risk or ameliorate progression of AD. The latest research, however, indicates that statins are not the solution for AD.
Cochrane reviews, considered the gold standard analysis of the best available information about healthcare interventions, focus primarily on randomized controlled trials (RCTs). In regards to the use of statins for AD prevention, reviewers evaluated the two large RCTs whose results have been published; neither showed any reduction in AD occurrence in patients treated with statins compared to those given placebo. Regarding treatment of AD, results from the large RCTs that have assessed this outcome have not been published, but initial analysis available from these studies indicates statins provide no benefit on the Alzheimer’s Disease Assessment Scale Cognitive Subscale.2
Not only do statins provide no benefit in preventing or treating AD, it appears they may do harm. A recently published analysis of results of a University of California-San Diego survey of 171 patients (age range 34-86 years) reporting statin-associated adverse cognitive effects, determined that cognitive ADRs were probably or definitely related to statin therapy in 128 patients (75%). Of 143 patients (84%) who reported stopping statin therapy, 128 (90%) reported improvement in cognitive problems. Median time to first-noted recovery was 2.5 weeks after statin discontinuation. Notably, in some patients, a diagnosis of dementia or Alzheimer’s disease was reversed. Nineteen patients whose symptoms improved or resolved after they discontinued statin therapy, and who then underwent rechallenge with a statin, exhibited cognitive problems again (multiple rechallenges with the same outcome in some).3
Given that, as discussed in Part I of this review, disordered cerebral cholesterol metabolism contributes to AD, why aren’t statins, HMG Co-A reductase inhibitors whose efficacy in aggressively lowering cholesterol is well documented, helpful in combating AD? A number of reasons are laid out in the research:
Cholesterol production typically decreases in AD brains. This is not a good thing! Cholesterol depletion has catastrophic consequences in neuronal culture preparations: it results in loss of vesicles and exocytotic activity, inhibits neurite growth and impairs neuronal survival. Evidence indicates that loss of cholesterol negatively and gravely impacts plasma membrane stability. In addition, the transduction of extracellular signals, essential for neuronal growth and survival, breaks down when levels of cholesterol become insufficient.4
When cholesterol is low, Abeta preferentially localizes at the membrane surface and assumes an aggregation-prone structure, but when the membrane contains ~33% cholesterol, the Abeta peptide inserts into the membrane with an altered structure that hinders peptide aggregation.5
Cholesterol is an essential component of the plasma membrane of all cells, including neurons, in which it increases plasma membrane rigidity and is found concentrated in lipid rafts together with sphingolipids and gangliosides (two classes of lipids that play important roles in cell signal transduction). In the adult, cholesterol plays key roles in the maintenance of neuronal plasticity and function. In lipid rafts, cholesterol acts as a spacer between sphingolipids and as a dynamic glue that holds the rafts together. Cholesterol removal results in raft disassembly with dissociation, deregulation and/or inactivation of most raft proteins.5
One of the proteins affected is plasmin, a raft-resident enzyme generated from an inactive precursor called plasminogen, whose activation occurs within lipid rafts. Plasmin cleaves Abeta peptides at multiple sites, thus preventing their aggregation, and also enhances APP cleavage by alpha-secretase. The lipid raft disorganization resulting from lowering neuronal membrane cholesterol prevents plasmin formation and activity, thus increasing production of Abeta peptides and oligomers.6
In the hippocampus of both humans and transgenic mice, only a very small amount of APP and beta-secretase (the enzyme that cleaves APP to produce Abeta) is found in the same membrane environment; with a moderate reduction of brain cholesterol, these co-localized APP and beta-secretase fractions increase markedly in rodents and in AD patients.7
Statins lower cholesterol by short-circuiting one of the earliest steps in cholesterol formation, the activity of HMGCoA reductase, thus statins’ action also blocks the synthesis of downstream intermediates with important physiological functions, including production of farnesylpyrophosphate, a precursor of molecules involved in cell signaling and inflammation, and ubiquinone (CoQ10), an antioxidant molecule that also plays a key role as an electron carrier in the mitochondrial electron transport chain. (In addition, via their inhibition of cholesterol, statins also short circuit synthesis of the entire class of steroid hormones, including DHEA and testosterone. A drop in either hormone may also contribute to loss of cognitive function.) Despite statins’ claim to anti-inflammatory effects in the vasculature, these actions promote oxidative stress and inflammation in the brain.
Another neuroprotective outcome of cholesterol synthesis in the brain occurs at the final step of the pathway in which desmosterol is reduced to cholesterol, a reaction catalyzed by the product of the Dhcr24 gene, also known as Seladin-1 (SELective Alzheimer’s Disease INdicator-1). In addition to participating in cholesterol synthesis, Seladin-1 has anti-apoptotic and neuroprotective effects against oxidative and oncogenic stimuli, and is directly involved in estrogen-mediated neuroprotection. Inhibition of cholesterol synthesis further down-regulates expression of Seladin-1, whose levels are already down-regulated in the AD brain.58
Finally, the cholesterol metabolites 24SOH-Chol and 27OH-Chol appear to regulate the activity of alpha-secretase and beta-secretase. 24SOH-Chol, the main cholesterol metabolite in the brain, reduces beta-secretase activity while increasing alpha-secretase activity.9
For all the above reasons, further lowering cholesterol and inhibiting its metabolism in the brain will promote rather than lessen Alzheimer’s disease, except possibly in one group of individuals—those who are carriers of the ApoE4 allele.
Statins Promote Insulin Resistance in ApoE2 and E3 Carriers, But May Benefit Those with ApoE4
It has also recently been revealed that statin users who are not carriers of the ApoE4 allele have greater insulin resistance (as indexed by greater HOMA-IR values) than ApoE4 carriers taking statins, or ApoE2 or ApoE3 carriers who are not statin users.10 These results suggest that statins could contribute to insulin resistance (and therefore risk of AD) in ApoE2 and ApoE3 carriers, but might provide protection against AD in ApoE4 carriers.
This relationship among statins, insulin resistance, and APOE genotype is likely related to the effects of each on lipid metabolism:
As noted in Part I of this review and immediately above, normal cerebral homeostasis is disrupted in AD. ApoE is a key cholesterol transport molecule in the periphery and the primary cholesterol carrier in the brain. Lipid efflux from neurons is significantly impacted by ApoE isoform. The ability of the ApoE2 and ApoE3 isoforms to induce lipid efflux, and therefore support normal cholesterol metabolism in the brain, is 2.5- to 3.9-fold greater than that of ApoE4. One of the key ways in which ApoE4 increases risk of AD is by reducing the rate at which cholesterol is recycled in the brain.11
In both the brain and the periphery, insulin is a primary regulator of lipid metabolism, stimulating lipogenesis and reducing lipolysis. In adipocytes, insulin resistance results in increased lipolysis. Thus insulin resistance produces increased levels of free fatty acids whose influx into the liver then inhibits insulin’s suppression of hepatic very low-density lipoprotein (VLDL) secretion, which normally quickly curtails post-prandial hyperlipidemia.
Insulin’s acute inhibitory effects on VLDL production allow the liver to rapidly restore levels of postprandial plasma lipids to an optimal physiologic range. In insulin-resistant individuals, however, insulin’s check on VLDL production is removed, so they experience higher and longer post-prandial increases of VLDL and other lipids with artery-damaging potential.
Epidemiological studies show increased peripheral lipid levels in mid-life can increase risk of AD 2- to 3-fold, which partly explains why ApoE4 carriers, who, even though they are less likely to be insulin resistant (as discussed in Part I of this review 12), have higher total and LDL cholesterol levels than non-ApoE4 carriers, and are at increased risk for AD.
In sum, statins may increase AD risk in non-ApoE4 carriers by reducing brain cholesterol levels as well as increasing insulin resistance in these individuals. Yet, in carriers of the ApoE4 allele, statins may reduce risk of AD by lowering peripheral total and LDL cholesterol in this genotype.
Don’t Be SAD, Tour the Mediterranean
In the last few years, research into the diet and nutrition relationship to disease has focused on dietary patterns instead of single nutrients for two key reasons: important interactions occur among components of a diet, and, more importantly, people do not eat isolated nutrients. Thus, researchers have developed dietary scores to estimate adherence to particular dietary patterns and their relationships to disease. One dietary pattern has consistently been associated with significantly reduced incidence of chronic degenerative diseases, including AD: the Mediterranean diet.13
Defining the Mediterranean Diet
First of all, “diet” in its typical connotation is a misnomer when applied to the Mediterranean diet, which is not a specific food regimen, but a collection of eating habits traditionally followed by people in the different countries bordering the Mediterranean Sea. Characterized by high consumption of vegetables, fruit, legumes, and complex carbohydrates (whole grains); moderate consumption of fish; the use of olive oil as the main source of fat; and low-to-moderate consumption of alcohol in the form of a little red wine with meals, this eating pattern has been reported to provide an optimal intake of antioxidant vitamins, polyunsaturated fats, and other beneficial nutrients for the prevention of chronic degenerative diseases, including AD.14
From the numerous recent studies investigating the association between nutrition and AD, the unsurprising discovery is that the Standard American Diet (SAD) promotes AD while a Mediterranean-style diet delivers neuroprotection against all forms of cognitive decline, including AD. The latest research confirming the Mediterranean diet’s cognitive benefits follows earlier studies indicating this dietary pattern is optimal for the prevention of key risk factors for AD, including cardio- and cerebro-vascular diseases (atherosclerosis, hypertension, stroke); disorders involving insulin resistance (e.g., MetS and type 2 diabetes); and obesity.
A recent meta-analysis involving 12 studies including a total of 1,574,299 subjects, followed for time periods ranging from 3 to 18 years, investigated the association between adherence to a Mediterranean diet and health status. The finding: greater adherence to a Mediterranean diet is associated with substantial improvement in health status, as indicated by significant reductions in overall mortality (9%), mortality from cardiovascular diseases (9%), incidence of or mortality from cancer (6%), and incidence of Parkinson’s disease and Alzheimer’s disease (13%).14
The Mediterranean Diet’s Anti-inflammatory / Anti-Alzheimer’s Effects
One reason for these healthy statistics, also discussed in the above meta-analysis, is that greater adherence to the Mediterranean diet is associated with beneficial effects on numerous inflammatory and coagulation markers (including significantly lower levels of homocysteine, C-reactive protein, interleukin-6, white blood cell count, and fibrinogen), lipids, and blood pressure – all important risk factors for cardiovascular diseases and for AD.
The Mediterranean diet’s anti-inflammatory effects have recently been further confirmed in a multicenter longitudinal study involving 1,003 myocardial infarction survivors from several European regions. In this study, each unit of increasing adherence to the “Mediterranean diet score” [further discussed below] correlated with a reduction of 3.1% in mean levels of C-reactive protein and of 1.9% in mean levels of interleukin-6.15
Homocysteine
Homocysteine serves as a good example of the ways in which the Mediterranean diet lessens AD risk. The presence of high levels of this intermediate compound of the methylation cycle is a common finding in patients affected by cognitive impairment and AD.1617
High homocysteine levels are associated with increased risk of cardiovascular and cerebrovascular ischemic diseases, which are well recognized risk factors for AD.18 Insufficiencies of B6, B12 or folic acid, all of which are necessary cofactors for homocysteine metabolism, have also been correlated with increased risk of cognitive decline and AD.18192021
In vitro studies have reported that homocysteine and deficiencies of folic acid impair DNA repair in hippocampal neurons, increasing susceptibility to toxicity from beta-amyloid protein.22 Homocysteine also potentiates the accumulation of soluble beta-amyloid in the brain, thus contributing to AD both indirectly via vascular damage and directly as a neurotoxin. Homocysteine is corrosive, degrading the main structural components of the artery: collagen and elastin.2425 In both the vasculature and brain, homocysteine and the product of its spontaneous oxidation, homocysteic acid, activate NMDA receptors, increasing intracellular levels of ionized calcium and ROS.26
The Mediterranean diet has been shown to significantly lower levels of homocysteine—an unsurprising outcome since this dietary pattern provides not only a variety of foods rich in B vitamins, but polyphenolic antioxidants, including resveratrol (in red wine and grape skins) and hydroxytyrosol (one of the main phenols in extra virgin olive oil), which have demonstrated protective effects against homocysteine-induced vascular damage.2728
Neuroprotective Fats
The brain is highly susceptible to oxidative damage because of its high metabolic rate and its abundance of oxidizable material, e.g., the polyunsaturated fatty acids that form the plasma membranes of neural cells. AD is characterized by a chronic inflammatory process around amyloid plaques, and increased levels of free radicals and pro-inflammatory cytokines are present in the brain of patients affected by AD.
Thus the SAD, with its typical pro-inflammatory omega-6:omega-3 ratio of ≥20:1, promotes AD, while the Mediterranean diet, which produces an anti-inflammatory omega-6:omega-3 ratio of 2:1 and has been shown to substantially lower the plasma ratio of omega-6 to omega-3 fatty acids within as little as 4 weeks, reduces oxidative stress and inflammation, lessening risk of AD.293031
Olive oil, the hallmark source of fat in the Mediterranean diet — albeit a monounsaturated fat (MUFA), whose main component (55-83%) is the omega-9 fatty acid, oleic acid, rather than a polyunsaturated fat (PUFA) — has also recently been associated with lowered risk of cognitive decline in a French three-city study involving 6,947 subjects followed for 4 years. Mechanistic explanations, in addition to the anti-inflammatory effects of olive oil phenols noted above, are provided by two recent papers: an article reporting that a virgin olive oil based breakfast repressed in vivo expression of several pro-inflammatory genes in mononuclear cells from patients with MetS , and the summary of the II international conference on olive oil and health. This consensus report references studies indicating that MUFA-rich membranes, although less fluid than those formed with PUFA, are also less vulnerable to oxidative damage, and suggests this translates to olive oil’s beneficial effects in maintaining the structural integrity of mitochondria and neuronal membranes, thereby promoting healthy neuronal transmission. 36
A Glass of Red Wine
Another hallmark of the Mediterranean diet is a small amount of alcohol in that daily glass of red wine enjoyed with meals. In addition to the resveratrol provided by red wine (which improves blood flow to the brain,37 and delivers protection against AD both via its direct antioxidant effects and via activation of sirtuin 138), elderly individuals who are not carriers of the ApoE4 allele also benefit from the light-to-moderate intake of alcohol, which is associated with a 45% reduction in AD risk.
Several papers have now reported likely molecular mechanisms for these benefits: in vitro, moderate ethanol preconditioning of hippocampal slices interferes with various glial-mediated neurotoxic responses to Abeta, and also causes an almost 3-fold increase in brain levels of heat shock protein 70, a protective molecular chaperone that inhibits induction of superoxide radical.404142
ApoE4 carriers can still benefit from phenolic-rich grape juice, which another recent paper indicates has comparable beneficial postprandial effects on endothelial function (specifically, flow mediated dilation) to red wine.43
Where the Tire Meets the Road—Estimating Patient Adherence
The adherence score seen in the research (originally proposed by Trichopoulou et al.44) was developed to enable researchers to estimate subjects’ global dietary pattern in relation to the typical characteristics of the Mediterranean diet. A value of 0 or 1 is assigned to each dietary component, whether protective/Mediterranean or potentially harmful/SAD. For each of the beneficial food groups (vegetables, fruits, legumes, whole grains, fish, moderate intake of red wine during meals, and the ratio of daily consumption of monounsaturated fats [from olive oil, nuts and seeds] to saturated fats), a value of 1 is assigned when the subject consumes more than the median of the population. A 0 value is assigned to individuals whose consumption is below the median. Concomitantly, for food categories considered detrimental (meat, especially red and processed meats, and full fat dairy products), a value of 1 is assigned to subjects whose consumption is below the median, and a 0 value is given to individuals whose consumption is above the median. The resulting score gives an indication of each individual’s adherence to the Mediterranean diet, which ranges from 0 (non-adherent) to 7-9 points (high adherence).
In your clinic, similar logic can be applied to help patients’ evaluate their adherence to a Mediterranean-type diet; however, since fewer than 1 in 10 Americans meet their calorie-specific MyPyramid fruit or vegetable recommendations (only 2.2% of men ≥19 years and 3.5% of women ≥19 years meet recommendations for fruit and vegetable consumption—a paltry 2.5 cups of vegetables and 2 cups of fruit in a 2,000 Kcal daily diet,{re45} being above the median will not suffice for prevention of AD. Thus following chart is an adaptation of the adherence score used in the research, and is provided as a simple aid in assisting patient awareness of the extent to which they are following a Mediterranean-type diet.
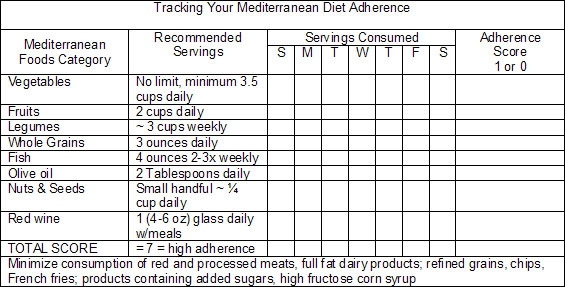
Mediterranean Diet Reduces Risk of Alzheimer’s Disease: Epidemiological Research
The possible association between the Mediterranean diet and reduced risk of AD is supported by a number of recent studies conducted by Scarmeas et al.{re44} In 2006, this group first published the results of their research on the effects of the Mediterranean diet and risk of AD in a community-based study involving 2,258 non-demented elders in New York, New York. Subjects were prospectively evaluated then followed for an average of 4 years. Higher adherence to the Mediterranean diet, i.e., being in upper third tertile of the adherence score, was associated with a 40% lower risk for AD.46
This result was confirmed by a further analysis of the data, a case-control study nested within the original cohort that investigated whether vascular risk factors (previous stroke, previous heart disease, diabetes, hypertension, lipids) lessened the protective effect of adherence to a Mediterranean diet. The positive association was found to be even stronger—a 68% reduction in AD risk among those in the highest tertile of adherence.47
In 2007, Scarmeas et al. published another paper in which they examined the association between adherence to the Mediterranean diet and mortality among the 192 subjects identified as having AD at the onset of the original study. Of these AD patients, 82 died during the following 4.4 years; however, patients with AD in the highest tertile of adherence to Mediterranean diet were 73% more likely to survive compared to those in the lowest tertile—a finding that strongly suggests adherence to a Mediterranean diet affects not only the risk of developing AD, but also the subsequent course of the disease.48
Additive Effect of Physical Activity and the Mediterranean Diet
The benefits of physical activity in reducing risk of cognitive decline (28% reduction in risk) and AD (45% reduction in risk) have been recently confirmed by a meta-analysis that included 16 prospective studies.49 But results are even better when physical activity is combined with adherence to the Mediterranean diet. Analyzing data gained from their prospective study of community-dwelling elders in New York, Scarmeas et al. found that physical activity plus higher adherence to Mediterranean diet have an additive effect. The combination results in a hazard ratio (HR) for AD of 35% versus an HR of 40% in those following a Mediterranean diet but engaging in little physical activity.50
Adherence to the Mediterranean diet has also been found to lessen risk of mild cognitive impairment—an early symptom of neurological dysfunction whose eventual outcome, if untreated, is likely to be AD, just as insulin resistance is an early warning sign of impending type 2 diabetes. Another study, again conducted by Scarmeas et al., identified an association between higher adherence to a Mediterranean diet and mild cognitive impairment in a population of 1,393 cognitively normal participants, 275 of whom developed mild cognitive impairment during 4.5 years of follow-up. Being in the upper third tertile of adherence to a Mediterranean diet was highly protective against developing mild cognitive impairment (and by implication AD), reducing risk by 48%.51
In their most recent paper, entitled “Food Combination and Alzheimer Disease Risk: A Protective Diet,” Scarmeas et al. evaluated dietary patterns in terms of their content of 7 potentially AD-related nutrients: saturated fatty acids, monounsaturated fatty acids, omega-3 polyunsaturated fatty acids, omega-6 polyunsaturated fatty acids, vitamin E, vitamin B12, and folate. What emerged was a dietary pattern strongly associated with lower AD risk. This dietary pattern was characterized by higher intakes of cruciferous vegetables, dark and green leafy vegetables, tomatoes, fruits, salad dressing, nuts, fish and poultry, and a lower intake of high-fat dairy products, red meat, organ meat, and butter. Sound familiar? Compared with subjects in the lowest tertile of adherence to this dietary pattern, the AD hazard ratio for subjects in the highest tertile was 0.62—i.e., a 38% reduction in risk for AD.52
Research presented at the Experimental Biology Meeting, held in Anaheim, Calif., April 26, 2010, also showed adherence to a Mediterranean diet slowed age-associated mental decline. Christy Tangney, PhD, and colleagues at Rush University Medical Center, followed 3,790 men and women in the Chicago Health and Aging Project (average age 75) for 7 years. Subjects answered a food-frequency questionnaire, spelling out in detail which components of the Mediterranean diet they ate and how often, and took a battery of tests every 3 years to evaluate mental functions, such as short- and long-term recall. Those with high adherence to the Mediterranean diet scored younger—the equivalent of 2 years younger than their age–in mental function. “The beauty of the finding,” Tangney is quoted as telling WebMD, “is that following the diet perfectly isn’t necessary to get a brain-protective effect. When someone incorporates a diet rich in fruits and vegetables and non-refined grains, such as cereals and breads, and breaks it up with a little wine, there appears to be at least some protection against cognitive aging.”53 Not too onerous a prescription.
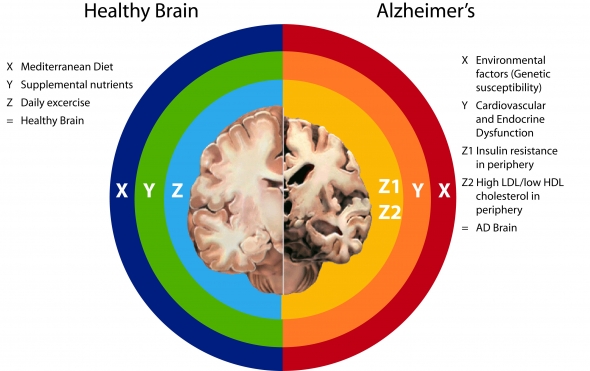
The Alzheimer’s Interstate (Right side of chart above)
(X) Environmental factors:
Standard American Diet: three problems—too much refined carbohydrate, saturated and trans fats AND pesticides, AND not enough micronutrients (e.g. insufficiencies of virtually all micronutrients, including vitamin A, vitamin D, vitamin K, omega-3s, B vitamins, & all the minerals including magnesium, calcium, zinc, selenium, potassium, etc.)
Couch Potato Lifestyle: combined with SAD exponentially promotes accumulation of visceral adipose tissue (VAT – belly fat), which is highly pro-inflammatory
Environmental toxins, e.g., pesticides (atrazine), BPA, POPs—all dysregulate endocrine function, promoting obesity, VAT, inflammation
Food intolerance, e.g., wheat, dairy, soy—provokes defensive immune response à inflammation
Genetic susceptibility, e.g., ApoE4
The above result in Cardiovascular and Endocrine Dysfunction as indicated by:
Unhealthy lipid profile: High Peripheral LDL Cholesterol / Low HDL Cholesterol levels
High Triglycerides
Hypertension (endothelial dysfunction)
Insulin resistance
Metabolic Syndrome
Diabetes
Obesity
(Y) Cardiovascular and Endocrine Dysfunctions promote:
Any and all of the above diseases/conditions promote Pro-inflamatory Metabolism, which causes à Leaky blood brain barrier à which opens up 2 pathways to AD:
- (Z1) Insulin resistance in periphery à hypometabolism in brainà increased production of ROS, RNS in periphery and brainà promotion beta-secretase activity à promotion Abeta formation àAD
- (Z2) High LDL/low HDL cholesterol in periphery à increased production of ROS, RNS in periphery à influx of Abeta-carrying LDL into the brain à dysregulation cholesterol homeostatis in brain à promotion beta-secretase activity à promotion Abeta formation àAD
End result: ↑ Alzheimer’s disease
Getting Off the AD Interstate (Left side of chart above)
Healthy Brain
(X) Mediterranean Dietàlow glycemic, low sat and trans-fat, high fiber, high omega-3, high micronutrients, e.g. B vitamins, myriad protective phenols (eg, resveratrol in red wine, vitamin E and hydroxytyrosol, tyrosol, and oleuropein in extra virgin olive oil)àall of which promote healthy lipid profile and good insulin sensitivityà a non-inflammatory metabolism
If avoid environmental toxicants (organic, use glass or stainless steel not BPA containing plastics) à less inflammation, less likelihood of endocrine dysruption
(Cautions—supply of vitamin A may not be adequate if one cannot convert carotenoids to retinol; supply of vitamin D may not be adequate depending on latitude and skin color)
(Y) Supplemental nutrientsà needed because: (1) a variety of SNPs result in increased need: SNPs have been identified for vitamin A, vitamin D (or, in the case of vitamin D, if a person has dark skin or lives in areas located at higher latitude), vitamin K, B vitamins. (2) because foods, especially conventionally grown foods, supply fewer micronutrients than they did 50 years ago, and (3) To restore mitochondrial efficiency in aging individuals, eg., CoQ10, N-acetyl cysteine, lipoic acid AND lastly, to help restore mitochondrial function in those experiencing brain hypometabolism — Medium chain triglycerides/ketones.
(Z) Daily exercise improves endothelial and mitochondrial function
The above support Optimal Cardiovascular and Endocrine Function as indicated by:
Healthy lipid profile: Total Cholesterol >200 / LDL > 130 / HDL ≤ 60
Triglycerides >150 mg/dL
Blood pressure >120/80 mm Hg
Insulin sensitive fasting insulin > 10 microU/mL
BMI within in range of 18.5 – 24.9
Normal cardiovascular and endocrine function promotes:
Anti-inflammatory metabolism à minimally permeable blood brain barrier
Insulin sensitivity and good endothelial function à normal glucose delivery and metabolism in the brain
Optimal cholesterol recycling / homeostasis in the brain
↑Alpha secreatase activity à ↑ neuroprotective APPsα
↓ Beta-secretase activity à ↓ Abeta
End Result: ↓ Alzheimer’s disease
The Spanish Ketogenic Mediterranean Diet
For those willing to go the extra dietary mile (or those at increased risk of AD, e.g., ApoE4 carriers), what is being called the Spanish Ketogenic Mediterranean Diet is likely to provide even more anti-AD bang for the dietary buck.54
Mildly ketogenic diets help preserve muscle mass, reduce appetite, favor increased fat loss, promote a non-atherogenic lipid profile, lower blood pressure and decrease insulin resistance. In contrast, it now appears that high carbohydrate diets, particularly when composed largely of refined carbohydrates (e.g., the SAD), are associated with: low levels of HDL; high levels of triglycerides, LDL and total cholesterol; MetS, type 2 diabetes, essential hypertension and cancer.54
In Spain, fish is an important protein source, thus the “Spanish Ketogenic Mediterranean Diet” whose effects were studied in 31 obese subjects, relied upon fish as the main source of protein, while using virgin olive oil as the principal source of fat and green vegetables and salads as the main source of carbohydrates, along with moderate red wine intake. Calories were unlimited, but subjects were encouraged to consume per day:
- A maximum of 30 grams of carbohydrates in the form of green vegetables and salads (2 portions of salad vegetables [such as alfalfa sprouts, lettuce, escarole, endive, mushrooms, radicchio, radishes, parsley, peppers, chicory, spinach, cucumber, chard and celery]), and 1 portion of low-carbohydrate vegetables (such as broccoli, cauliflower, cabbage, artichoke, eggplant, squash, tomato and onion). Allowable salad dressings were olive oil, vinegar, lemon juice, garlic, salt, herbs and spices.
- A minimum of 30 ml (=1 ounce or 2 tablespoons) of virgin olive oil distributed as 10 ml (2 teaspoons) at breakfast, lunch and dinner.
- 200–400 ml (6-12 ounces) of red wine distributed as 100-200 ml (6-8 ounces) at lunch and dinner.
- Unlimited protein in the form of fish 4 days each week (all types except longer-lived predators, swordfish and shark due to concerns re mercury), and meat, fowl, eggs, shellfish and cheese on the remaining 3 days.
- Trans-fats and processed meats with added sugar were not allowed.
- No more than two cups of coffee or tea and at least 3 litres of water (~12 8-oz glasses) were recommended daily.
While for the average American patient, the Spanish ketogenic diet borders on the draconian, at least some might be willing if given the data on this dietary regime’s highly impressive beneficial impacts on numerous risk factors for AD. These included significant reductions in body weight (108.62 kg–> 94.48 kg), BMI (36.46 kg/m(2)–>31.76 kg/m(2), systolic blood pressure (125.71 mmHg–>109.05 mmHg), diastolic blood pressure (84.52 mmHg–> 75.24 mmHg), total cholesterol (208.24 mg/dl–>186.62 mg/dl), triglycerides (218.67 mg/dl–>113.90 mg/dl—a 47.91% reduction), glucose (109.81 mg/dl–> 93.33 mg/dl), and LDL (114.52 mg/dl–>105.95 mg/dl), and a significant increase in HDL (50.10 mg/dl–>54.57 mg/dl).
Toning Up the AD Brain with Ketones
Exceptional results, such as those seen in the preceding study, are the driving force behind growing research interest in ketones’ potential as protective agents against AD, particularly in carriers of ApoE4.
As noted in Part I of this review, a decline in brain glucose metabolism is seen long before significant amounts of Abeta are present, years before the onset of frank AD (even as early as age 30 in ApoE4 carriers), and brain hypometabolism is a consistent pathological feature of AD.
Unlike other tissues in the body, the brain does not efficiently metabolize fats, instead relying almost exclusively on glucose as its energy substrate. Thus, chronic cerebral glucose hypometabolism can profoundly affect brain function, and some researchers believe dysfunctional energy metabolism not only precedes but is key to alterations in APP processing that favor Abeta production.5556
Dysfunctional alterations in lipid/glucose metabolism may be triggered by:
- Environmental factors, e.g., exposure to the commonly used herbicide, atrazine. Recently published animal research demonstrated that chronic exposure to extremely low levels of atrazine decreased basal metabolic rate, and increased body weight, intra-abdominal fat and insulin resistance without changing food intake or physical activity level. (Reason to suggest your patients eat organic!)57
- The standard American diet. High in refined carbohydrates, the SAD inhibits the use of fatty acids, increases triglyceride levels,58 and is also very low in omega-3 fatty acids. The latter results in low levels of DHA in the brain. Insufficient brain levels of DHA promote beta-secretase cleavage of APP, and the presence of less flexible fats in neural plasma membranes may also result in impaired functioning of other proteins key for glucose metabolism, such as glucose transporters.59
- Genetic predisposition, e.g., possession of an APOE4 allele. The ApoE4 protein increases triglyceride levels by binding triglycerides much more readily than ApoE2 or 3, while also inhibiting lipolysis.58
Supplementing the normal glucose supply of the brain with ketone bodies (i.e., acetoacetate, beta-hydroxybutyrate, and acetone), which are normally produced from fat stores when glucose supplies are limited, e.g., during prolonged fasting or a ketogenic diet, has demonstrated significant benefit in both animal models of neurodegenerative disorders and in human clinical trials, including trials involving patients with AD.60
However, although the ketogenic diet has demonstrated beneficial effects on dyslipidemia and insulin resistance, and specifically in AD, research on ketones and epilepsy indicates that to produce therapeutic levels of ketone bodies, 90% of calories must come from fat. Obviously, such a diet is impractical for chronic use because a regimen this high in fat and low in carbohydrate is unpalatable; compliance will be poor. For this reason, a way in which to obtain high ketone levels, while allowing the patient to eat a relatively normal diet, has been under investigation. Medium chain triglycerides may offer a solution.60
Reversing AD-Promoting Brain Hypometabolism with Medium Chain Triglycerides
To date, only one study has been published evaluating the effectiveness of ketone bodies in treating human AD subjects. This study utilized medium chain triglycerides (MCT), unique triglycerides comprised of fatty acid chains between 5 to 12 carbons. Because of their short fatty acid chain lengths, MCT are not treated like long chain fatty acids, but undergo obligate oxidation. If sufficient MCT are ingested, the excess acetyl-CoA produced will generate ketone bodies. Also, importantly, the oxidation of MCT occurs regardless of other macronutrients consumed; thus, MCT use escapes the carbohydrate restrictions necessary for ketone production in a ketogenic diet.
A crossover study examined the effects of acute elevation of serum ketone bodies on cognitive performance in 20 mild to moderate probable AD subjects. A single, 40 gram dose of MCT induced a 10-fold elevation in ketone bodies (specifically, beta-hydroxybutyrate) after 2 hours. Ninety minutes after dosing, subjects were tested for changes in cognitive performance. The single administration of MCT led to a significant correlation between performance on the Alzheimer’s Disease Assessment Scale-Cognitive subscale (ADAS-Cog), a paragraph recall test, and serum beta-hydroxybutyrate concentration, with those subjects presenting the highest beta-hydroxybutyrate levels showing the most improvement. The rapid (90 minute) improvement suggests that the effect is a result of improved neuronal metabolism.61
Also, in this study, subjects who were not carriers of ApoE4 showed greater improvement in ADAS-Cog scores than those who were ApoE4 carriers, and this outcome has been replicated in a longer, 90-day dosing study.62
Why would MCT work preferentially in subjects who are not carriers of APOE4? Suzanne Craft, PhD, whose research is discussed in Part I of this review, has suggested that differences in insulin sensitivity resulting from Apo E4 carriage status are responsible. Non-carriers of ApoE4 have been shown to have higher fasting insulin levels and lower glucose disposal rates than normal controls, suggesting insulin resistance.63
The insulin resistance of non-ApoE4 AD subjects may also explain their responsiveness to ketone bodies, which are transported into the brain by the monocarboxylate transporter (MCTs) carrier proteins, one of which (MCT1) is widely expressed and found in endothelial cells of the blood brain barrier (BBB). BBB Levels of monocarboxylate transporters are elevated in diabetes and in other conditions in which insulin resistance occurs, such as in the hypometabolic state that precedes and accompanies AD in the brain.
A clinically relevant point: the serum level of ketone bodies reached in the above study is easily achievable, requiring only a single 40 gram dose of MCT, and thus individuals at risk or suffering from AD may gain the benefits of ketosis without draconian dietary changes. However, the research clearly indicates that one cannot remain on the SAD, but must move to the Mediterranean diet to prevent or mitigate progression of AD.
MCTs are naturally present in coconut oil and red palm kernel oil. MCT oil derived from these sources is also available.
From A to Zinc—Optimal Nutrition, the Best Hedge vs. Alzheimer’s?
As triage theory, the latest hypothesis generated by legendary researcher Bruce Ames, PhD, suggests, nutrients are triaged to meet the body’s most pressing survival needs. For example, under scarcity conditions, vitamin K is preferentially shunted to its use in the clotting cascade as K1, rather than converted to K2 in the intestines and used to activate the matrix Gla protein that prevents arterial calcification or carboxylate osteocalcin to build bone.6465
Even from this necessarily truncated review, it should be apparent that sufficiency of virtually all nutrients needed for optimal lipid and glucose metabolism, and mitochondrial function, plays a role in AD prevention—how important a role being related to an individual’s genetic inheritance of single nucleotide polymorphisms (SNPs). For example, several B vitamin SNPs have been identified that result in higher than RDI need for B6, B12 and folate, all of which are necessary for methylation / homocysteine metabolism, and for choline, which is an essential component of acetylcholine. What this means is that a significant number of individuals are functionally deficient even if consuming the RDI levels of these nutrients—and we know that few Americans are getting RDI levels from the SAD.66
Oxygen, although considered free currency, has also been found to be a conditionally essential nutrient in many brain disorders.6768 Chronic stress causes transient cerebral hypoxia,69 which leads to expression of iNOS in the brain, causing microglial activation and increased risk for AD. Stress reduction via physical exercise and meditation may thus provide significant protective benefit against AD. Hypoxia is also associated with hypertension, implying the necessity of healthy endothelial function, which is supported by L-arginine. For a review of L-arginine’s effects on endothelial function, please see: “Longevity Medicine Strategies for Cardiovascular Disease: Closing the Statin Gap in Endothelial Dysfunction and Insulin Resistance Naturally, with L-Arginine and Citrulline: Part I and Part II”
Heavy metal toxicity promotes AD. Both aluminum and mercury bind to thyroid hormone receptors and prevent proper metabolism. In addition to chelation, iodine is needed, both to displace heavy metals and to produce thyroid hormone, which also necessitates selenium. Thyroid hormone regulates APP gene expression. Hypothyroidism is common in the elderly, and the reduced action of thyroid hormone on the APP gene contributes to AD pathology by increasing APP expression*, brain hypometabolism, ROS, RNS, and thus increased levels of Abeta. (*Specifically, T3 represses APP promoter activity by binding to the nuclear T3 receptor (TR). The unliganded receptor increases promoter activity; the effect of T3 binding is to return promoter activity to basal levels.71)
While one could argue that insufficiency of virtually any nutrient contributes to AD pathology, the most recent research identifies a couple with major impact: vitamins A and D.
Two Vital Anti-Alzheimer’s Micronutrients
“A” Does Not Stand for Alzheimer’s
Vitamin A plays key roles in alpha-secretase production, acetylcholine transmission, and the regulation/inhibition of excessive microglial activation.
Two binding sites for the retinoic acid receptor (RXR) are found just 203 and 302 nucleotides upstream of the gene for the most prevalent alpha-secretase, ADAM-10, and, not coincidentally, vitamin A, specifically retinoic acid (RA), has been shown to significantly upregulate ADAM-10 mRNA levels.7273In cell cultures, direct application of RA to hippocampal slices from vitamin A deficient mice reverses impairment of ADAM-10 transcription and upregulates production of both ADAM-10 and APP, resulting in reduced Abeta formation and increased formation of APPsα, which has neurotrophic and neuroprotective properties. In adult rats, dietary deficiency of vitamin A results in Abeta deposition in cerebral blood vessels.7475
In humans, vitamin A insufficiency has been identified as a contributing factor in late-onset AD (LOAD), the form of the disease that afflicts 98% of those affected.767778A number of potential mechanisms, not simply RA’s effect on alpha-secretase production, are likely responsible.
In addition to its impact on alpha-secretase, RA insufficiency downregulates production of acetylcholine transferase, thus inhibiting neurotransmission of acetylcholine, whose impaired release is another hallmark of AD.79
Retinoic acid may also help prevent AD via numerous anti-inflammatory effects, which combine to inhibit microglial activation. RA strongly suppresses production of IL-6, a key pro-inflammatory cytokine, and also inhibits amyloid-beta-induced production of TNF-alpha, and thus the expression of inducible NO synthase (iNOS), in microglia—effects that are thought to be mediated via retinoids’ inhibition of NF-kappaB nuclear translocation.79
As discussed in the LMR review Vitamin A – Tolerance Extends Longevity, RA is involved in numerous anti-inflammatory activities that promote the development of regulatory T cells, which tune down the inflammatory TH1/TH17 immune response and increase the production of anti-inflammatory cytokines, e.g. 1L-10.
Lastly, it is has just recently been recognized that the vitamin A metabolite, retinol, not only functions as the precursor for retinaldehyde (the universal chromophore in the vertebrate and invertebrate eye) and RA, but itself plays a fundamental role in the mitochondria. Retinol is an essential cofactor for protein kinase C delta, and the PKC-delta/retinol complex signals the pyruvate dehydrogenase complex to increase influx of pyruvate into the Krebs cycle. When cells are deprived of retinol, respiration and ATP synthesis defaults to basal levels. Thus, vitamin A serves as a nutritional sensor with a key role in regulating mitochondrial energy homeostasis.80
Vitamin A insufficiency may be a key contributing factor, to the hypometabolism seen in the AD brain. Since reduced cerebro-glucose metabolism can be seen years before AD diagnosis (as noted by AD-researcher Suzanne Craft and discussed in Part I of this review), vitamin A supplementation may help correct this underlying dysfunction and prevent progression to AD.
The recognition of even one of the above facets of vitamin A activity would identify impaired retinoid metabolism as significant in AD. Fortunately, it is one that can be easily resolved by ensuring vitamin A sufficiency. The potentially good news is that, since vitamin A insufficiency is much more common than has been previously thought, it may be a key contributing – and rectifiable — factor in a large percentage of those afflicted with AD.
Vitamin A insufficiency can result not only from insufficient consumption of foods rich in pre-formed vitamin A (liver is the best source of vitamin A; egg yolk, cow’s milk, butter, cheese and fatty fish provide some) or pro-vitamin A (aka beta-carotene, which is concentrated in carrots, sweet potato, kale, spinach, winter squash, collard greens, cantaloupe, mango), but to recently identified genetic risk factors. Two common SNPs (present in ~25-40% of the population) result in very poor conversion of carotenoids to retinoic acid. For full discussion of these issues plus additional factors that can result in retinoic acid insufficiency, please see our LMR article: Common Genetic Variants and Other Host-related Factors Greatly Increase Susceptibility to Vitamin A Deficiency. For an in-depth discussion of vitamin A’s effects on immunity, relationship to vitamin D, and recommendations regarding assessment of patients’ vitamin A status, dosage and safety issues, please see our review: Vitamin A — Tolerance Extends Longevity.
Vitamin D: Illuminating the Brain’s Golden Years with Supplemental Sunshine
Vitamin D insufficiency correlates with increased risk for AD. In a study of 318 elders receiving home care from 2003 to 2007, vitamin D [25(OH)D3, aka calcidiol] insufficiency (≤20 ng/mL) was associated with more than twice the odds of all-cause dementia (odds ratio [OR] = 2.3), Alzheimer disease (OR = 2.5) and stroke (with and without dementia symptoms) (OR = 2.0).81
Examination of a cross-sectional group of 80 participants, 40 with mild AD and 40 non-demented persons selected from a longitudinal study of memory and aging, found vitamin D deficiency (<20 ng/mL) was associated an 11.69 odds ratio for an active mood disorder and significantly impaired cognitive performance.82
A number of studies have also revealed comorbidity of AD and osteoporosis, and a recent paper discussed the finding that blood biomarkers of osteoporosis (C-terminal collagen fragments and osteocalcin) are significantly increased in AD patients.8384
Vitamin D’s anti-inflammatory effects, which in the brain include down-regulating microglial activation, may provide one mechanism for this connection. Inflammation activates osteoclasts, and thus plays an important role in osteoporosis. The AD brain exhibits significant inflammation, with microglial activation as the driving force provoking its inflammatory cascade.858687
Abeta protein activates microglia, which produce ample quantities of peroxynitrite and other oxidants, cytokines, and pro-inflammatory prostaglandins that hit neurons with a double destructive whammy: they boost Abeta production and induce apoptosis. Peroxynitrite also increases neural sensitivity to excitotoxicity, and potentiates the direct toxicity of Abeta to neurons.
Activated microglia may also adversely impact neuron function and survival less directly by impairing astrocyte function. Healthy astrocytes sequester glutamate, thus protecting neurons by alleviating excitotoxicity. Peroxynitrite and microglial-derived cytokines have been shown to impair glutamate uptake by astrocytes. Furthermore, peroxynitrite and PGE2 (a prostaglandin derived from the omega-6 fatty acids rampant in the standard American diet) increase neurons’ susceptibility to excitotoxic cell death.88
Microglial activation also induces expression of the 1-alpha-hydroxylase that converts 25(OH)D3 to its active hormonal form. Microglial cells express the vitamin D receptor, and the binding of 1,25(OH)D3 (aka calcitriol) suppresses the expression of iNOS by microglial cells exposed to inflammatory activators. (The research has used lipopolysaccharide (LPS), an endotoxin that induces a strong immune response and to which humans are particularly sensitive.)
In addition, calcitriol boosts astrocyte production of a protective neural growth factor called glial-derived neurotrophic factor (GDNF), which protects dopaminergic neurons of the substantia nigra. 1α-Hydroxylase, the enzyme that converts 25(OH)D3 (calcidiol) to the active hormone, 1,25(OH)D3 (calcitriol), is only expressed by activated, but not quiescent, microglia, and activated microglia generate calcitriol when incubated with 25(OH)D3, suggesting invocation of a self-regulatory inhibition of the inflammatory cascade—if sufficient vitamin D is present.
Conclusion
While the research evidence rarely, if ever, reaches a “the sun is likely to rise tomorrow” level, surely the evidence presented in this review provides rational grounds to argue that the fear, pessimism and victim mentality espoused by the NIH consensus panel on AD prevention is unfounded, and worse, may exert a nocebo effect on patient health outcomes. We now have a level of insight into the underlying causes of AD pathology that reveals late-onset AD is largely due to diet and lifestyle practices, which are modifiable and highly responsive to dietary and supplement interventions.89
Please read Part I: Alzheimer’s and Atherosclerosis – Siblings in a Dysfunctional Family: Part I
References
- Panel: No Evidence of Alzheimer’s Prevention. Webmd.com. ↑
http://www.webmd.com/alzheimers/news/20100428/panel-no-evidence-alzheimers-prevention - McGuinness B, Passmore P. J . Can Statins Prevent or Help Treat Alzheimer’s Disease?. Alzheimers Dis. 2010 Feb 24. ↑
Abstract - Evans MA, Golomb BA. Statin-associated adverse cognitive effects: survey results from 171 patients. Pharmacotherapy. 2009 Jul;29(7):800-11. ↑
Abstract - Pfrieger FW. Role of cholesterol in synapse formation and function. Biochim Biophys Acta. 2003 Mar 10;1610(2):271-80. ↑
Abstract - Stefani M, Liguri G. Cholesterol in Alzheimer’s disease: unresolved questions. Curr Alzheimer Res. 2009 Feb;6(1):15-29. ↑
Abstract - Ledesma MD, Abad-Rodriguez J, Galvan C, et al. Raft disorganization leads to reduced plasmin activity in Alzheimer’s disease brains. EMBO Rep. 2003 Dec;4(12):1190-6. ↑
Abstract - Abad-Rodriguez J, Ledesma MD, Craessaerts K, et al. Neuronal membrane cholesterol loss enhances amyloid peptide generation. J Cell Biol. 2004 Dec 6;167(5):953-60. ↑
Abstract - Ledesma MD, Dotti CG. The conflicting role of brain cholesterol in Alzheimer’s disease: lessons from the brain plasminogen system. Biochem Soc Symp. 2005;(72):129-38. ↑
Abstract - Famer D, Meaney S, Mousavi M, et al. Regulation of alpha- and beta-secretase activity by oxysterols: cerebrosterol stimulates processing of APP via the alpha-secretase pathway. Biochem Biophys Res Commun. 2007 Jul 20;359(1):46-50. ↑
Abstract - Vanfossen BT, Watson GS, Baker LD, et al. Statin Users Without an APOE-epsilon4 Allele have Increased Insulin Resistance. J Alzheimers Dis. [Epub ahead of print] . ↑
Abstract - Minagawa H, Gong JS, Jung CG, et al. Mechanism underlying apolipoprotein E (ApoE) isoform-dependent lipid efflux from neural cells in culture. J Neurosci Res. 2009 Aug 15;87(11):2498-508. ↑
Abstract - Craft S. The role of metabolic disorders in Alzheimer disease and vascular dementia: two roads converged. Arch Neurol. 2009 Mar;66(3):300-5. ↑
Abstract - Sofi F. The Mediterranean diet revisited: evidence of its effectiveness grows. Curr Opin Cardiol. 2009 Sep;24(5):442-6. ↑
Abstract - Sofi F, Cesari F, Abbate R, et al. Adherence to Mediterranean diet and health status: meta-analysis. BMJ. 2008 Sep 11;337:a1344. ↑
Abstract - Panagiotakos DB, Dimakopoulou K, Katsouyanni K, et al. Mediterranean diet and inflammatory response in myocardial infarction survivors. Int J Epidemiol. 2009 Jun;38(3):856-66. Epub 2009 Feb 24 . ↑
Abstract - de Toledo Ferraz Alves TC, Ferreira LK, et al. Cardiac Disorders as Risk Factors for Alzheimer’s Disease. J Alzheimers Dis. 2010 Apr 22. [Epub ahead of print] . ↑
Abstract - Rosendorff C, Beeri MS, Silverman JM. Cardiovascular risk factors for Alzheimer’s disease. Am J Geriatr Cardiol. 2007 May-Jun;16(3):143-9. ↑
Abstract - Clarke R, Smith AD, Jobst KA, et al. Folate, vitamin B12, and serum total homocysteine levels in confirmed Alzheimer disease. Arch Neurol. 1998 Nov;55(11):1449-55. ↑
Abstract - Tucker KL, Qiao N, Scott T, et al. High homocysteine and low B vitamins predict cognitive decline in aging men: the Veterans Affairs Normative Aging Study. Am J Clin Nutr. 2005 Sep;82(3):627-35. ↑
Abstract - Kado DM, Karlamangla AS, Huang MH, et al. Homocysteine versus the vitamins folate, B6, and B12 as predictors of cognitive function and decline in older high-functioning adults: MacArthur Studies of Successful Aging. Am J Med. 2005 Feb;118(2):161-7. ↑
Abstract - Teunissen CE, Blom AH, Van Boxtel MP, et al. Homocysteine: a marker for cognitive performance? A longitudinal follow-up study. J Nutr Health Aging. 2003;7(3):153-9. ↑
Abstract - Kruman II, Kumaravel TS, Lohani A, et al. Folic acid deficiency and homocysteine impair DNA repair in hippocampal neurons and sensitize them to amyloid toxicity in experimental models of Alzheimer’s disease. J Neurosci. 2002 Mar 1;22(5):1752-62. ↑
Abstract - Seshadri S, Wolf PA. Homocysteine and the brain: vascular risk factor or neurotoxin?. Lancet Neurol. 2003 Jan;2(1):11. ↑
Abstract - Vizzardi E, Bonadei I, Zanini G, et al. Homocysteine and heart failure: an overview. Recent Pat Cardiovasc Drug Discov. 2009 Jan;4(1):15-21. ↑
Abstract - Hubmacher D, Cirulis JT, Miao M, et al. Functional consequences of homocysteinylation of the elastic fiber proteins fibrillin-1 and tropoelastin. J Biol Chem. 2010 Jan 8;285(2):1188-98. ↑
Abstract - Boldyrev AA. Molecular mechanisms of homocysteine toxicity. Biochemistry (Mosc) . 2009 Jun;74(6):589-98. ↑
Abstract - De Lorenzo A, Noce A, Bigioni M, et al. The effects of Italian Mediterranean organic diet (IMOD) on health status. Curr Pharm Des. 2010;16(7):814-24. ↑
Abstract - Carluccio MA, Ancora MA, Massaro M, et al. Homocysteine induces VCAM-1 gene expression through NF-kappaB and NAD(P)H oxidase activation: protective role of Mediterranean diet polyphenolic antioxidants. Am J Physiol Heart Circ Physiol. 2007 Oct;293(4):H2344-54. ↑
Abstract - Manios Y, Detopoulou V, Visioli F, et al. Mediterranean diet as a nutrition education and dietary guide: misconceptions and the neglected role of locally consumed foods and wild green plants. Forum Nutr. 2006;59:154-70. ↑
Abstract - Ambring A, Johansson M, Axelsen M, et al. Mediterranean-inspired diet lowers the ratio of serum phospholipid n-6 to n-3 fatty acids, the number of leukocytes and platelets, and vascular endothelial growth factor in healthy subjects. Am J Clin Nutr. 2006 Mar;83(3):575-81. ↑
Abstract - Sofi F, Macchi C, Abbate R, et al. Effectiveness of the Mediterranean Diet: Can It Help Delay or Prevent Alzheimer’s Disease? . J Alzheimers Dis. 2010 Feb 24. [Epub ahead of print] . ↑
Abstract - Wärnberg J, Gomez-Martinez S, Romeo J, et al. Nutrition, inflammation, and cognitive function. Ann N Y Acad Sci. 2009 Feb;1153:164-75. ↑
Abstract - Dauncey MJ. New insights into nutrition and cognitive neuroscience. Proc Nutr Soc. 2009 Nov;68(4):408-15. Epub 2009 Aug 24. ↑
Abstract - Berr C, Portet F, Carriere I, et al. Olive oil and cognition: results from the three-city study. Dement Geriatr Cogn Disord. 2009;28(4):357-64. ↑
Abstract - Camargo A, Ruano J, Fernandez JM, et al. Gene expression changes in mononuclear cells from patients with metabolic syndrome after acute intake of phenol-rich virgin olive oil. BMC Genomics. 2010 Apr 20;11(1):253. [Epub ahead of print] . ↑
Abstract - López-Miranda J, Pérez-Jiménez F, Ros E, et al. Olive oil and health: summary of the II international conference on olive oil and health consensus report, Jaén and Córdoba (Spain) 2008. Nutr Metab Cardiovasc Dis. 2010 May;20(4):284-94. Epub 2010 Mar 19. ↑
Abstract - Kennedy DO, Wightman EL, Reay JL, et al. Effects of resveratrol on cerebral blood flow variables and cognitive performance in humans: a double-blind, placebo-controlled, crossover investigation. Am J Clin Nutr. 2010 Mar 31. [Epub ahead of print] . ↑
Abstract - Sun AY, Wang Q, Simonyi A, et al. Resveratrol as a Therapeutic Agent for Neurodegenerative Diseases. Mol Neurobiol. 2010 Mar 21. [Epub ahead of print] . ↑
Abstract - Luchsinger JA, Tang MX, Siddiqui M, et al. Alcohol intake and risk of dementia. J Am Geriatr Soc. 2004 Apr;52(4):540-6. ↑
Abstract - Belmadani A, Kumar S, Schipma M, et al. Inhibition of amyloid-beta-induced neurotoxicity and apoptosis by moderate ethanol preconditioning. Neuroreport. 2004 Sep 15;15(13):2093-6. ↑
Abstract - Collins MA, Neafsey EJ, Mukamal KJ, et al. Alcohol in moderation, cardioprotection, and neuroprotection: epidemiological considerations and mechanistic studies. Alcohol Clin Exp Res. 2009 Feb;33(2):206-19. ↑
Abstract - Collins MA, Neafsey EJ, Wang K, et al. Moderate Ethanol Preconditioning of Rat Brain Cultures Engenders Neuroprotection Against Dementia-Inducing Neuroinflammatory Proteins: Possible Signaling Mechanisms. Mol Neurobiol. 2010 Apr 28. [Epub ahead of print] . ↑
Abstract - Hampton SM, Isherwood C, Kirkpatrick VJ, et al. The influence of alcohol consumed with a meal on endothelial function in healthy individuals. J Hum Nutr Diet. 2010 Jan 22. [Epub ahead of print] . ↑
Abstract - Trichopoulou A, Costacou T, Bamia C, et al. Adherence to a Mediterranean diet and survival in a Greek population. N Engl J Med. 2003 Jun 26;348(26):2599-608. ↑
Abstract - Kimmons J, Gillespie C, Seymour J, et al. Fruit and vegetable intake among adolescents and adults in the United States: percentage meeting individualized recommendations. Medscape J Med. 2009;11(1):26. Epub 2009 Jan 26. ↑
Abstract - Scarmeas N, Stern Y, Tang MX, et al. Mediterranean diet and risk for Alzheimer’s disease. Ann Neurol. 2006 Jun;59(6):912-21. ↑
Abstract - Scarmeas N, Stern Y, Mayeux R, et al. . Mediterranean diet, Alzheimer disease, and vascular mediation. Arch Neurol. 2006 Dec;63(12):1709-17. ↑
Abstract - Scarmeas N, Luchsinger JA, Mayeux R, et al. Mediterranean diet and Alzheimer disease mortality. Neurology. 2007 Sep 11;69(11):1084-93. ↑
Abstract - Hamer M, Chida Y. Physical activity and risk of neurodegenerative disease: a systematic review of prospective evidence. Psychol Med. 2009 Jan;39(1):3-11. Epub 2008 Jun 23. ↑
Abstract - Scarmeas N, Luchsinger JA, Schupf N, et al. Physical activity, diet, and risk of Alzheimer disease. JAMA. 2009 Aug 12;302(6):627-37. ↑
Abstract - Scarmeas N, Stern Y, Mayeux R, et al. Mediterranean diet and mild cognitive impairment. Arch Neurol. 2009 Feb;66(2):216-25. ↑
Abstract - Gu Y, Nieves JW, Stern Y, Luchsinger JA, Scarmeas N. Food Combination and Alzheimer Disease Risk: A Protective Diet. Arch Neurol. 2010 Apr 12. [Epub ahead of print] . ↑
Abstract - Mediterranean Diet May Save Brain Power. Webmd.com. ↑
http://www.webmd.com/healthy-aging/news/20100427/mediterranean-diet-may-save-brain-power - Pérez-Guisado J, Muñoz-Serrano A, Alonso-Moraga A. Spanish Ketogenic Mediterranean Diet: a healthy cardiovascular diet for weight loss. Nutr J. 2008 Oct 26;7:30. ↑
Abstract - Craft S. The role of metabolic disorders in Alzheimer disease and vascular dementia: two roads converged. Arch Neurol. 2009 Mar;66(3):300-5. Review. ↑
Abstract - Henderson ST. High carbohydrate diets and Alzheimer’s disease. Med Hypotheses. 2004;62(5):689-700. ↑
Abstract - Lim S, Ahn SY, Song IC, Chung MH,et al. Chronic exposure to the herbicide, atrazine, causes mitochondrial dysfunction and insulin resistance. PLoS One. 2009;4(4):e5186 . ↑
Abstract - Hellerstein MK. Carbohydrate-induced hypertriglyceridemia: modifying factors and implications for cardiovascular risk. Curr Opin Lipidol. 2002 Feb;13(1):33-40. ↑
Abstract - Calon F, Cole G. Neuroprotective action of omega-3 polyunsaturated fatty acids against neurodegenerative diseases: evidence from animal studies. Prostaglandins Leukot Essent Fatty Acids. 2007 Nov-Dec;77(5-6):287-93. ↑
Abstract - Henderson ST. Ketone bodies as a therapeutic for Alzheimer’s disease. Neurotherapeutics. 2008 Jul;5(3):470-80. ↑
Abstract - Reger MA, Henderson ST, Hale C. Effects of beta-hydroxybutyrate on cognition in memory-impaired adults. Neurobiol Aging. 2004 Mar;25(3):311-4. ↑
Abstract - Henderson et al. Manuscript in preparation cited in Henderson ST. Ketone bodies as a therapeutic for Alzheimer’s disease. Neurotherapeutics. 2008 Jul;5(3):470-80. ↑
Abstract - Craft S, Asthana S, Schellenberg G, et al. Insulin effects on glucose metabolism, memory, and plasma amyloid precursor protein in Alzheimer’s disease differ according to apolipoprotein-E genotype. Ann N Y Acad Sci. 2000 Apr;903:222-8. ↑
Abstract - Ames BN. Low micronutrient intake may accelerate the degenerative diseases of aging through allocation of scarce micronutrients by triage. Proc Natl Acad Sci U S A. 2006 Nov 21;103(47):17589-94. Epub 2006 Nov 13. Review. ↑
Abstract - McCann JC, Ames BN. Vitamin K, an example of triage theory: is micronutrient inadequacy linked to diseases of aging? . Am J Clin Nutr. 2009 Oct;90(4):889-907. Epub 2009 Aug 19. Review. ↑
Abstract - Ames BN. Optimal micronutrients delay mitochondrial decay and age-associated diseases. Mech Ageing Dev. 2010 Apr 23. [Epub ahead of print] . ↑
Abstract - Peña F, Ramirez JM. Hypoxia-induced changes in neuronal network properties. Mol Neurobiol. 2005 Dec;32(3):251-83. ↑
Abstract - Jan Marino Ramirez, PhD, interviewed by Jeff Bland, PhD, . Functional Medicine Update. Vol. 30, No. 2(February 2010 issue), distributed by Synthesis, available at http://www.jeffreybland.com/. ↑
http://www.jeffreybland.com/ - Khovryakov AV, Podrezova EP, Kruglyakov PP, et al. Involvement of the NO synthase system in stress-mediated brain reactions. Neurosci Behav Physiol. 2010 Mar;40(3):333-7. Epub 2010 Feb 13. ↑
Abstract - O’Barr SA, Oh JS, Ma C, et al. Thyroid hormone regulates endogenous amyloid-beta precursor protein gene expression and processing in both in vitro and in vivo models. Thyroid. 2006 Dec;16(12):1207-13. ↑
Abstract - Belandia B, Latasa MJ, Villa A, et al. Thyroid hormone negatively regulates the transcriptional activity of the beta-amyloid precursor protein gene. J Biol Chem. 1998 Nov 13;273(46):30366-71. ↑
Abstract - Tippmann F, Hundt J, Schneider A, et al. Up-regulation of the alpha-secretase ADAM10 by retinoic acid receptors and acitretin. FASEB J. 2009 Jun;23(6):1643-54. ↑
Abstract - Koryakina A, Aeberhard J, Kiefer S, et al. Regulation of secretases by all-trans-retinoic acid. FEBS J. 2009 May;276(9):2645-55. ↑
Abstract - Corcoran JP, So PL, Maden M. Disruption of the retinoid signalling pathway causes a deposition of amyloid beta in the adult rat brain. Eur J Neurosci. 2004 Aug;20(4):896-902. ↑
Abstract - Husson M, Enderlin V, Delacourte A, et al. Retinoic acid normalizes nuclear receptor mediated hypo-expression of proteins involved in beta-amyloid deposits in the cerebral cortex of vitamin A deprived rats. Neurobiol Dis. 2006 Jul;23(1):1-10. ↑
Abstract - Fahrenholz F. Alpha-secretase as a therapeutic target. Curr Alzheimer Res. 2007 Sep;4(4):412-7. ↑
Abstract - Goodman AB, Pardee AB. Evidence for defective retinoid transport and function in late onset Alzheimer’s disease. Proc Natl Acad Sci U S A. 2003 Mar 4;100(5):2901-5. Epub 2003 Feb 25. ↑
Abstract - Lane MA, Bailey SJ. Role of retinoid signalling in the adult brain. Prog Neurobiol. 2005 Mar;75(4):275-93. ↑
Abstract - Shudo K, Fukasawa H, Nakagomi M, et al. Towards retinoid therapy for Alzheimer’s disease. Curr Alzheimer Res. 2009 Jun;6(3):302-11. ↑
Abstract - Acin-Perez R, Hoyos B, Zhao F, et al. Control of oxidative phosphorylation by vitamin A illuminates a fundamental role in mitochondrial energy homoeostasis. FASEB J. 2010 Feb;24(2):627-36. ↑
Abstract - Buell JS, Dawson-Hughes B, Scott TM, et al. 25-Hydroxyvitamin D, dementia, and cerebrovascular pathology in elders receiving home services. Neurology. 2010 Jan 5;74(1):18-26. Epub 2009 Nov 25. ↑
Abstract - Wilkins CH, Sheline YI, Roe CM, et al. Vitamin D deficiency is associated with low mood and worse cognitive performance in older adults. Am J Geriatr Psychiatry. 2006 Dec;14(12):1032-40. ↑
Abstract - Luckhaus C, Mahabadi B, Grass-Kapanke B, et al. Blood biomarkers of osteoporosis in mild cognitive impairment and Alzheimer’s disease. J Neural Transm. 2009 Jul;116(7):905-11. Epub 2009 May 26. ↑
Abstract - Tysiewicz-Dudek M, Pietraszkiewicz F, Drozdzowska B. Alzheimer’s disease and osteoporosis: common risk factors or one condition predisposing to the other? . Ortop Traumatol Rehabil. 2008 Jul-Aug;10(4):315-23. ↑
Abstract - Lue LF, Kuo YM, Beach T, et al. Microglia Activation and Anti-inflammatory Regulation in Alzheimer’s Disease. Mol Neurobiol. 2010 Mar 3. [Epub ahead of print] . ↑
Abstract - Tuohimaa P, Keisala T, Minasyan A, et al. Vitamin D, nervous system and aging. Psychoneuroendocrinology. 2009 Dec;34 Suppl 1:S278-86. ↑
Abstract - McCann JC, Ames BN. Is there convincing biological or behavioral evidence linking vitamin D deficiency to brain dysfunction? . FASEB J. 2008 Apr;22(4):982-1001. Epub 2007 Dec 4. ↑
Abstract - McCarty MF. Toward prevention of Alzheimers disease–potential nutraceutical strategies for suppressing the production of amyloid beta peptides. Med Hypotheses. 2006;67(4):682-97. Epub 2006 Jul 7. ↑
Abstract - McCarty MF. Down-regulation of microglial activation may represent a practical strategy for combating neurodegenerative disorders. Med Hypotheses. 2006;67(2):251-69. Epub 2006 Mar 2. ↑
Abstract
Comments are closed.