
Abstract
Amyloid precursor protein (APP) is the transmembrane protein that, if cleaved by the enzyme beta-secretase, produces amyloid beta (the histopathological hallmark of Alzheimer’s disease [AD]). If acted upon by the enzyme alpha-secretase, however, APP cleavage produces neuroprotective compounds. What determines which enzyme will gain access to APP? The answers to this question reveal connections among cardiovascular disease, neurological dysfunction and nutrient insufficiencies that provide the rationale for new therapeutic strategies to prevent AD.
Part I of this review discusses recent evidence that Alzheimer’s disease and atherosclerosis are siblings. Amyloid plaque and atherosclerotic plaque have much in common. Both are damage control responses to high cholesterol in a pro-inflammatory environment, the result, in many individuals, of insulin resistance. Amyloid beta (Abeta) plays an apolipoprotein-like role in helping to maintain cerebral cholesterol homeostasis, and Abeta peptides are found in atherosclerotic plaque. In both brain and arteries, soluble Abeta peptides upregulate inducible nitric oxide synthase (iNOS), leading to increased production of reactive nitrogen species (RNS) and reactive oxygen species (ROS). Exacerbated ROS/RNS production results in macrophage activation in the arteries and glial activation in the brain, both of which promote beta-secretase processing of APP and Abeta formation—a potentially vicious cycle. In contrast, alpha-secretase and the metabolic conditions that promote its activation and access to APP—insulin sensitivity, good mitochondrial and endothelial function; a healthy lipid profile, and key micronutrients, including the lead actors necessary for oxidative phosphorylation, vitamin D and retinoic acid—are highly neuroprotective.
Part I of this review summarizes the latest research, which reveals that AD is the ultimate outcome in the brain of systemic vascular and glycemic dysregulation. Awareness of what goes awry promotes optimism along with new understanding of the mechanisms underlying dietary and micronutrient modulation, the topic of Part II of our review. The causes of AD are neither mysterious nor untreatable: Alzheimer’s disease is not the inescapable fate of the aging brain.
Amyloid Precursor Protein Processing: Promoting or Preventing Alzheimer’s Disease
Amyloid precursor protein (APP) is a transmembrane protein concentrated in the plasma (outer) membrane of brain cells, including neurons, glial cells, the cells lining the perivascular channels that drain into the CNS, and the endothelial cells of the blood brain barrier. Not only is APP one of the most abundant proteins present in the CNS, it is also ubiquitously expressed in peripheral tissues including muscles, epithelial tissue, and circulating cells. Platelets represent the most important peripheral source of APP and contain APP concentrations equivalent to those found in brain.1
APP can be cleaved and processed by either alpha- or beta-secretase enzymes. In both cases, the initial cleavage is followed by an additional cleavage by gamma-secretase. All tissues and cells containing APP, including platelets, also contain alpha, beta-secretase and gamma-secretases. Thus, Abeta can be formed and stored within platelets, then released upon platelet activation. (Not only are platelets essential for primary hemostasis and repair of the endothelium, they also play a key role in the development of atherosclerotic plaques; platelets are a source of inflammatory mediators, and platelet activation by inflammatory triggers is a significant contributing factor in cerebrovascular disease.2
Alpha-secretase is protective not only because it cleaves APP inside the amylolid beta (Abeta) sequence, splitting this sequence apart and thus preventing the formation of Abeta peptides, but also because it releases a soluble fragment called APPsα that is neuroprotective. Beta-secretase cleavage frees the entire Abeta sequence, producing Abeta peptides which, if not quickly cleared when produced in or delivered to the brain, become potent neurotoxins that ultimately self-aggregate and deposit as amyloid plaque.3
A substantial amount of research has been conducted on ADAM-10, a member of the ADAM family of proteins (ADAM =a disintegrin and metalloproteinase) that, among other functions, acts as an alpha-secretase and has been shown to prevent amyloid plaque formation in an Alzheimer’s disease mouse model.4 In mice bred to over-express APP in neurons, concomitant over-expression of ADAM-10 causes increased production of APPsα, reduced formation of amyloid plaque, and a reduction in cognitive deficits. In contrast, in mice bred to possess an inactive ADAM-10, its lack results in Alzheimer-like pathology.5
Levels of alpha-secretase (ADAM-10) and APPsα are reduced in the cerebral spinal fluid (CSF) of Alzheimer’s disease (AD) patients compared to controls. Thus, researchers have begun to ask, “What lessens alpha-secretase production or activity, and what might increase it?” Surprising discoveries appearing in the evolving research on APP and Abeta provide potentially therapeutic answers.
Be forewarned: many new pieces of the puzzle are emerging. What may, in the following discussion, initially appear to be disparate topics will meld together by this article’s end to present a deeper understanding of Alzheimer’s pathophysiology that enables proactive, effective prevention.
Abeta: Good Intentions, Gone Awry
Abeta, a readily self-aggregating peptide that produces severe neurotoxicity, particularly in its initial form as a soluble monomer or oligomer*, is eventually deposited in brain tissue interstices in plaques, which, recently styled “the cobwebs of the brain,” are the histopathological hallmarks of AD.6 (*It is now generally recognized that mature amyloid fibrils/plaques are virtually devoid of toxicity to cells; it is their unstable precursors, the oligomers or prefibrillar aggregates of Abeta peptides, that are extremely toxic.)7 For a detailed discussion of this topic, please see the LMR review titled: Alzheimer’s Disease: a 21st Cenury Epidemic.
Although Abeta is widely known only for its infamous role as the driving pathological persona in AD, seemingly invented by Nature in order to ruin the aging brain, the impetus for its increased cerebral production is cholesterol homeostasis. New research shows that Abeta’s physiological raison d’etre is to help recycle cholesterol within the brain as well as promote its efflux to the systemic circulation. Increased Abeta production is part of a protective response through which the brain attempts to lower its cellular cholesterol levels.
As noted above, Abeta is produced from amyloid precursor protein (APP) in a sequential process in which APP is cleaved first by beta- and then gamma-secretases, enzymes whose activity is upregulated by high levels of cholesterol – one of many reasons why high cholesterol (particularly in individuals carrying the ApoE4 genotype, which binds more avidly to Abeta and is also pro-inflammatory8) is a risk factor for Alzheimer’s disease.
Abeta increases cholesterol efflux from cells, assembling high-density lipoprotein-like particles during its secretion. In animal studies, lipoproteins with Abeta are excreted into peripheral tissues much more efficiently than those without Abeta.9 Thus, Abeta is not simply a neuro-terrorist. Abeta plays an apolipoprotein-like role in the maintenance of cerebral cholesterol homeostasis.
Increasing cholesterol in the plasma membrane of brain cells increases cerebral Abeta production
Cholesterol-rich lipid rafts are platforms where APP clusters in the plasma membrane (the outer cell membrane) of neurons, astrocytes and microglia. When cholesterol concentration decreases in the plasma membrane, so does Abeta production from APP, while neuroprotective alpha-secretase activity goes up. When cholesterol concentrations increase in the plasma membrane, alpha-secretase activity drops, and beta-secretase activity and Abeta production increase.
Recently, researchers unexpectedly found that increasing plasma membrane cholesterol caused endosomes in hippocampal neuronal cells to enlarge by 31%.10 (Endosomes are organelles in which molecules are sorted on their way to lysosomes for degradation.) Enlarged endosomes are seen in affected neurons in the brains of AD patients and develop years before either of the neuropathological hallmarks of AD, Abeta plaque and tau hyperphosphorylation, become evident.
The appearance of enlarged endosomes coincides with increased production of Abeta peptides, suggesting that increased plasma membrane cholesterol causes endosomal pathology that increases APP processing by beta-secretase, resulting in Abeta overproduction. This pathology tends to become self-perpetuating since endosome enlargement is also induced by the C-terminal fragment of APP, which is the first item produced en route to Abeta formation when APP is cleaved by beta-secretase.11
Therapeutic news to use: the amyloidogenic pathway is initiated to lower brain cholesterol levels
A large body of evidence now indicates that increased levels of plasma membrane cholesterol promote amyloidogenic processing of APP. Cholesterol normally concentrates in the plasma membrane of neurons and glial cells. The immediate precursor to Abeta, the C-terminal domain of APP (called C99 because it consists of a domain containing 99 residues), is the product of beta-secretase cleavage of APP and the substrate of gamma-secretase cleavage, from which Abeta issues. It now appears that this section of APP has a propensity to bind with cholesterol.
Such binding favors the amyloidogenic pathway because it promotes localization of APP/C99 to lipid rafts in the plasma and endosome membranes, where beta and gamma-secretase are concentrated. On the other hand, non-amyloidogenic alpha-secretase is believed to reside primarily in the bulk membrane (the non-raft/ free cholesterol sections of the plasma membrane) and to be inactivated when forced to associate with lipid rafts.12
Cholesterol within the plasma and endosome membranes is distributed between the two pools: free cholesterol in the bulk membrane and cholesterol associated with lipid rafts. At low plasma membrane/endosomal cholesterol concentrations, free cholesterol and free APP/C99 will predominate, and APP will primarily be subject to alpha-secretase-initiated processing. However, as cholesterol levels in the membrane rise, lipid rafts will predominate. APP/C99 will complex with cholesterol in these rafts, and will be processed by beta- and gamma-secretase.
Research suggests that the second and third products in the amyloidogenic pathway—Abeta and the intracellular domain of the APP (AICD)—act to lower cellular cholesterol levels. It has been shown that Abeta stimulates the release of cholesterol and some other lipids from cells in the form of lipoproteins, and also that Abeta fibrills down-regulate cholesterol biosynthesis.
Other studies suggest that Abeta reduces biosynthesis of cholesterol and other lipids under conditions of ischemia, and that cytosolic Abeta acts as an inhibitor of HMG-CoA reductase, the rate-limiting enzyme in the biosynthetic pathway for cholesterol whose activity is blocked by statins. (Statins, however, are not the answer to the cholesterol issues in AD. The reasons why are discussed in Part II of this review.)
In addition, the AICD, which is released when gamma-secretase cleaves C99 to produce Abeta, translocates to the cell nucleus, where it acts as a transcriptional suppressor to the gene that encodes the LRP1 protein. LRP1 is a major apoE receptor in the brain that mediates cellular cholesterol uptake via endocytosis (the process by which cells absorb molecules from outside the cell by engulfing them with their cell membrane). Thus AICD may also help down-regulate cellular cholesterol uptake.
In sum, Abeta can down-regulate cerebral cholesterol content. All these observations suggest that the amyloidogenic pathway’s true function is to reduce total cellular cholesterol levels. Further, the fact that APP now appears to be a cholesterol binding protein suggests that one of its raisons d’etre may be to act as a cellular cholesterol sensor/receptor. When membrane cholesterol levels are elevated, APP forms a complex with cholesterol, which promotes the amyloidogenic pathway, whose products then reduce both cholesterol uptake and biosynthesis, completing a negative-feedback loop.12
Amyloidogenic and Non-Amyloidogenic APP Processing
Flowchart by John Morgenthaler
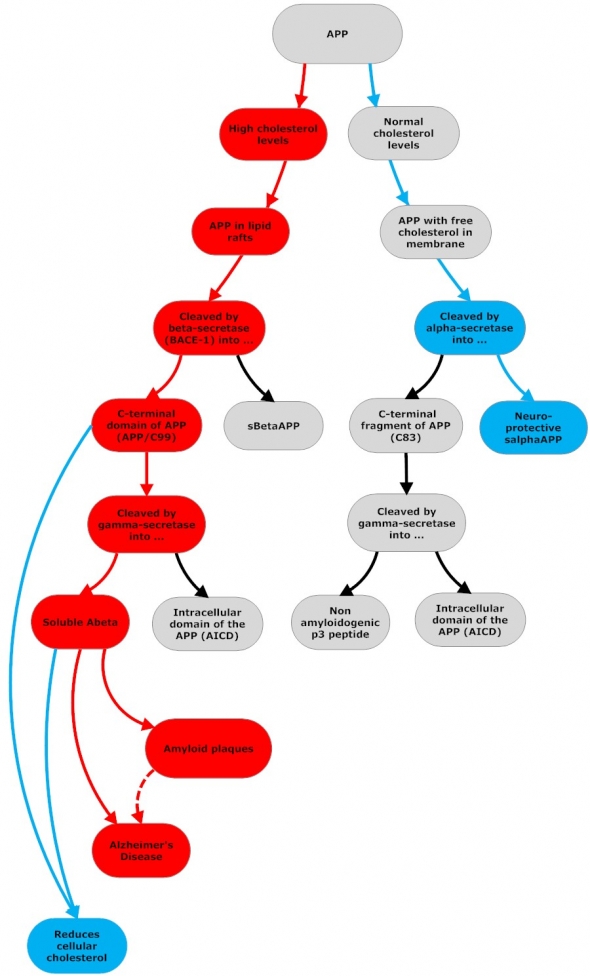
APP can be cleaved and processed by either alpha- or beta-secretase enzymes. In both cases, the initial cleavage is followed by an additional cleavage by gamma-secretase. Beta-secretase cleavage produces C99 and APPsβ. C99 is then cleaved again by gamma-secretase and releases Abeta and AICD(APP intracellular domain). Abeta and AICD lower cellular cholesterol levels. (1) Abeta stimulates cholesterol efflux from cells in the form of HDL-like particles in which Abeta is contained; (2) Abeta fibrils (cytosolic Abeta) down-regulate cholesterol biosynthesis by inhibiting HMG-CoA reductase (3) AICD translocates to the cell nucleus and suppresses the gene that encodes LRP1, which mediates cellular cholesterol uptake.
Amyloid Beta and the Cholesterol Connection to Alzheimer’s Disease
In the brain, persistently elevated cholesterol promotes excessive Abeta production, the formation of neurotoxic souble Abeta oligomers, and the deposition of amyloid plaque. When cholesterol levels are within optimal range, alpha-secretase is the constitutive enzyme, APP is channeled to non-amyloidogenic processing, and Abeta is produced in lesser amounts and assists in cholesterol recycling and efflux, helping to maintain cholesterol homeostasis.4 When cholesterol levels remain high, however, Abeta quickly metamorphoses from Dr. Jekyll to Mr. Hyde.
Risk Factors Shared by Alzheimer’s & Cardiovascular Disease
- APOE e4 allele: ApoE is involved in the binding and clearance of brain Abeta; ApoE2 and ApoE3 are much more effective than apoE4, which is also associated with higher systemic cholesterol levels. ApoE4 promotes a pro-inflammatory and immuno-reactive phenotype, while the other two isoforms, particularly ApoE2, are protective vs. oxidative stress.
- Hypertension: both causes and results in endothelial dysfunction, iNOS production, increased oxidative stress, cerebral hypoxia
- Hypercholesterolemia / Dyslipidemia: lipid profile showing not only increased levels of total plasma cholesterol & LDL but reduced levels of HDL
- Decreased plasma apolipoprotein A-1 (apoA-I): the major protein component of HDL, apoA-1 promotes cholesterol efflux. High serum apoA-1 is linked to decreased risk of coronary artery disease; also to significantly lower risk of dementia if not carrying an ApoE4 allele.
- Increased ApoB: the primary apolipoprotein in LDL, binds to LDL receptors and delivers cholesterol, also binds Abeta peptide; ApoB levels correlate with Abeta deposition
- Insulin resistance / MetS / Type 2 Diabetes
- Obesity
- Vascular dementia and LOAD also share numerous risk factors including hypertension, dyslipidemia and diabetes.
Location, location, location
The alpha- and beta-secretases concentrate in different areas of the cell. Not surprisingly, given beta-secretase’s cholesterol efflux function, this enzyme is primarily found in the Golgi apparatus and lysosomes. The Golgi is an organelle in most eukaryotic cells that serves as the processing, packaging center for macromolecules, e.g., proteins and lipids, which pass through the Golgi soon after their synthesis before making their way to their destination. When brain metabolism is functioning well, Abeta’s next destination is the endosome, a further sorting compartment from which Abeta is sent on to the lysosomes, the disposal unit of the cell, for degradation. Also not surprisingly, beta-secretase has optimal activity at the acidic pH found within the endosomes. As noted above, beta-secretase is also active in “lipid rafts” in the plasma membrane.1314
In contrast, alpha-secretase activity is optimized in cell surface membrane regions with few lipid rafts, especially areas with high fluidity where this secretase is primarily localized, another reason for the importance of the most fluid of all fats, the omega-3 fatty acid, DHA, in promoting a-secretase activity and cognitive function.15 For a detailed discussion of this topic, please see the LMR review titled Omega-3s, ApoE Genotype and Cognitive Decline.
Thus, if APP accumulates at the cell surface, it has a much better chance of being cleaved by alpha-secretase. If APP is shifted to inside the cell to the Golgi, endosomes or lysosome compartments where beta-secretase is the key player, then processing to Abeta will be the likely outcome. Emerging evidence indicates that cholesterol and the family of low-density lipoprotein receptors (LDLR) impact where APP will accumulate.16 And so does insulin, which significantly accelerates APP trafficking from the Golgi to the plasma membrane (more on insulin’s connections to Abeta and AD below in the section titled Insulin Resistance: the Dysglycemic Route to Atherosclerosis & Alzheimer’s below).17
APP Location Chaperones – the Low-Density Lipoprotein Receptors
At least four members of the low-density lipoprotein receptor (LDLR) family regulate APP location: LRP (LDLR-related protein), LRP1B, SorLA/R11 (Sorting protein related receptor containing LDLR class A repeats) and apoER2 (apoE receptor 2). LRP and apoER2 promote delivery of APP to areas rich in beta-secretase; LRP1B and SorLA/R11 deliver APP to alpha-secretase-rich areas.16 (More on these chaperones, which work with ApoE, below.)
Cholesterol dysregulation in the brain: key cause of Abeta overload and AD
Although it accounts for only 2% of the body mass, the brain contains 25% of the total body amount of unesterified (free) cholesterol; over 99% of the cholesterol in the brain is unesterified. Brain tissue is separated from blood circulation and other tissues by the blood–brain barrier, (although this barrier is turning out to not be nearly as impermeable as formerly thought)7 and its cholesterol is almost completely synthesized de novo by glial cells. Brain lipids, including cholesterol, are primarily located in cell plasma membranes and are constantly being replaced.18
Brain cholesterol is essential for a wide variety of key brain functions, and when released from degenerating nerve terminals, is actively recycled by glial cells and astrocytes, esterified by cholesterol esterase, then cleared from the extracellular matrix by binding, primarily to ApoE, which delivers it to neurons. A negative feedback loop between free cholesterol, Abeta and HMG-CoA reductase maintains intracellular cholesterol equilibrium in the brain. In the AD brain, however, this orderly process of cholesterol turnover is impaired.
Although the brain synthesizes its own cholesterol and does not depend on the circulation for its cholesterol supply, it has recently been shown that, contrary to previously accepted opinion, plasma lipoproteins do cross the blood brain barrier (BBB). In individuals in whom cholesterol levels are persistently (or even frequently post-prandially) elevated, plasma lipoproteins can deliver significant amounts of cholesterol (and the Abeta it contains) to the CNS.7
A key role for ApoE
AD can be classified broadly into two groups, early onset AD (EOAD, occurring < 65 years) and late-onset AD (LOAD, occurring > 65 years). EOAD, although the most severe form, accounts for only a small percentage of AD cases (<5%), and the majority of these are caused by mutations in one of three genes: those for APP, presenilin (PS) 1 and PS2.
LOAD accounts for the vast majority of AD cases (>95%). Age and possession of the ApoE4 single nucleotide polymorphism (SNP) are considered the major risk factors for LOAD. ApoE4 is also a major risk factor for CVD, due not only to its association with increased low density lipoprotein levels, but also higher oxidative stress, and a more pro-inflammatory / immunoreactive phenotype.19 For more on this topic, please see our LMR article: Omega-3s, ApoE Genotype and Cognitive Decline
In the adult brain, astrocytes not only synthesize, but also internalize and recycle the cholesterol released from degenerating nerve terminals. This recycled cholesterol is then delivered back to neurons, an activity that requires cholesterol’s binding to one of the variants of apolipoprotein E (ApoE), which serves as the major lipoprotein in the CNS. ApoE is a ligand for cell surface lipoprotein receptors (including the APP chaperones noted above); is responsible for regulating lipid transport among brain cells; and clears cholesterol from the extracellular space, delivering it to the lysosomes for recycling.
Abeta is internalized in ApoE-containing particles, which then move either to endosomal compartments in the plasma membrane, from which they are sent to lysosomes for recycling, or efflux from the brain into perivascular channels that drain into the CNS and the systemic circulation. Once in the systemic circulation, they are sent to and processed by the liver, where Abeta peptides are catabolized and excreted into the bile.8
The ApoE4 isoform has the highest affinity for Abeta binding, but is the least effective ApoE isoform in promoting cholesterol efflux (from macrophages as well as from neurons and astrocytes); the most immunoreactive isoform; and has the least antioxidant capacity – all reasons ApoE4 increases risk for AD).13 If these ApoE4 traits were not problematic enough, recent papers indicate that the ApoE4 variant also promotes Abeta-induced apoptosis in neuronal cells. After actively binding Abeta, ApoE4 forms a reactive molecular intermediate with the potential to insert into the lysosomal membrane, destabilize it, and cause lysosomal leakage, provoking apoptosis.2021
Arterial Abeta Leads to Leaky Brain and Dysregulation of Cerebral Cholesterol Homeostasis
While the intact BBB normally largely prevents cholesterol import into the CNS from the peripheral circulation, vascular injury can enable cholesterol-carrying lipoproteins to gain relatively easy access to the CNS. This results in increased brain membrane cholesterol, increased beta-secretase cleavage of APP, and increased generation of Abeta peptides. In addition, oxidized cholesterol derivatives found in the circulating blood can also cross the BBB. Hence, any vascular damage (e.g., the endothelial damage seen in atherosclerosis, hypertension, MetS, diabetes) can result in increased importation of damaged cholesterol into the brain.2021 This is where systemic, not just cerebral, endothelial dysfunction enters the AD causality chain.
Plus, Abeta is also found in platelets inside human atherosclerotic plaques. In vitro studies have shown that when perivascular macrophages phagocytose platelets within atherosclerotic plaques, APP is released and processed to Abeta-like peptides, which then promote the upregulation of inducible NO-synthase (iNOS). iNOS activation results in increased production of toxic amounts of NO, causing additional RNS/ROS production, additional macrophage activation, endothelial dysfunction, and the onset of a vicious cycle.2223
Recent data indicate that by enhancing iNOS production, Abeta peptides in atherosclerotic plaque promote vasoconstriction that ultimately reduces cerebral blood flow and increases the production of superoxide by NADPH oxidase, which is located in both vascular and brain cell membranes. The outcomes: damage to the vascular endothelium, reduced brain perfusion and cerebral ischemia, increased production of iNOS, glial activation, mitochondrial dysfunction – a brain-destructive Pachinko game.
Arterial and Amyloid Plaque: Different Location, Same Theme
Altered cholesterol metabolism results in failure to clear Abeta and a chronic inflammatory response by the microglia, the brain’s version of macrophages, which are surprisingly similar to those found in the intima of atherosclerotic lesions. Brain microglia produce virtually all the same cytokines, chemokines, growth factors, enzymes, complement and coagulation factors, and ROS, as their vascular counterparts, plus the microglia express many of the same surface receptors that mediate local immune reactions. In addition to microglia, both astrocytes and neurons, also swing into action, producing inflammatory mediators, including C-reactive protein, amyloid P, and complement factors.13 All of this contributes to increased production of ROS and RNS, endothelial dysfunction, mitochondrial dysfunction, hypoxia—sound familiar?—many roads to Rome, all converging on Alzheimer’s disease.
In what other research also suggests is the brain’s variation on atherosclerosis, Abeta plays a role similar in many respects to that undertaken by marcophages in the vasculature. Macrophages ingest and attempt to sequester oxidized LDL, becoming foam cells and furthering an inflammatory process whose end result is the formation of arterial plaque. Similarly, Abeta, failing to sufficiently lower cholesterol in the brain (where highly active neuronal mitochondria greatly increase the potential for damage by ROS/RNS), sequesters excess cholesterol in amyloid plaque. Different location, same theme.13
Convergent Pathways to Alzheimer’s Disease: High Systemic Cholesterol &/or Insulin Resistance
Flowchart by John Morgenthaler

In both brain and arteries, soluble Abeta peptides upregulate inducible nitric oxide synthase (iNOS), leading to increased production of reactive nitrogen species (RNS) and reactive oxygen species (ROS). Whether induced by high cholesterol or insulin resistance, exacerbated ROS/RNS production in the systemic vasculature results in the activation of platelets (the most important peripheral source of APP, containing APP concentrations equivalent to those found in brain) and macrophages in the arteries and glial activation in the brain, both of which promote beta-secretase processing of APP and Abeta formation—a potentially vicious cycle.
Insulin Resistance: the Dysglycemic Route to Atherosclerosis & Alzheimer’s
Although the human brain represents only 2% of the body weight, it utilizes 15% of the cardiac output, 20% of total body oxygen consumption, and 25% of total body glucose.24 Because the energy-hungry brain relies on glucose as its only source of fuel for ATP production, (except during fasting or significant carbohydrate restriction when a switch is made to ketone metabolism), insulin plays a pivotal role in cognition and other aspects of normal brain function.*
Insulin resistance results in lowered insulin activity and chronically elevated levels of insulin in the periphery, but a reduction in insulin levels in the brain. Knowing this, it is not surprising that dysfunctions related to systemic insulin resistance — metabolic syndrome (MetS, aka insulin resistance syndrome) and related conditions such as type 2 diabetes mellitus, hypertension and dyslipidemia — are all also associated with age-related cognitive decline and AD.26
In addition to depriving the brain of insulin (and therefore its glucose fix), insulin resistance promotes an increase in the production of inflammatory agents, including Abeta. When plasma insulin is experimentally increased in healthy humans to the high levels typically seen in patients with insulin resistance, levels of inflammatory agents and Abeta increase in the brain.27
Insulin and Abeta have a reciprocal relationship
Insulin regulates Abeta in several ways:
By increasing its trafficking from within to outside the cell. When high levels of insulin are given, Abeta levels go up in spinal fluid. Insulin crosses the BBB, enters the CNS, and promotes trafficking of Abeta out into extracellular space in the brain, which then drains into the spinal fluid. While this is a positive effect of insulin, it’s also a double-edged sword. Abeta needs to be outside the cell to get degraded, but if not degraded quickly, soluble Abeta peptides are highly neurotoxic. Enter “insulin-degrading enzyme” to the rescue.28
By increasing the expression and therefore availability of insulin-degrading enzyme. As its name implies, insulin-degrading enzyme (IDG) degrades insulin, but it is also responsible in the brain for degrading Abeta. Insulin thus impacts Abeta levels both by competing for IDG, so supplies become insufficient to clear Abeta, and by regulating levels of IDG in the brain since insulin increases the expression and therefore availability of IDG.28
Abeta that manages to bind to dendrites fights back by causing the neurons’ insulin receptors to move off the neuronal surface and into the cell body, where they are no longer accessible to insulin.2829 On the positive flip side, restoration of insulin sensitivity down regulates the availability of Abeta binding sites, thus preventing pathogenic Abeta oligomers from binding.30
The Insulin Connection to Alzheimer’s Disease
Flowchart by John Morgenthaler

Insulin resistance/chronically elevated levels of insulin in the periphery correlates with a reduction in cerebro-glucose metabolism that can be seen years before a diagnosis of Alzheimer’s disease and results in impaired mitochondrial energy production (more ROS, RNS) and increased Abeta production. Insulin regulates Abeta in several ways. When insulin crosses the blood brain barrier, its presence in the brain increases the expression of insulin-degrading enzyme, which also degrades Abeta. Insulin promotes Abeta trafficking into the extracellular space in the brain where it can be degraded by insulin-degrading enzyme or drain into the spinal fluid, be effluxed into the systemic circulation and delivered to the liver for removal. Insulin also down-regulates the expression of Abeta binding sites. In reciprocal fashion, when Abeta binds to dendrites it promotes insulin resistance by causing the neurons’ insulin receptors to move off the cell surface and into the cell body, where they are no longer accessible to insulin.
Many tributaries, two main streams, one pathological outcome
It has recently been proposed by Suzanne Craft and her research team at the University of Washington School of Medicine, Seattle, WA, that there are two main pathways to AD, one of which is driven by physiological processes associated with the ApoE4 allele, while the second is driven by insulin resistance.31
According to Dr. Craft, if you take a group of patients with AD, about 50% will have the ApoE4 allele, a much higher percentage of ApoE4 carriers than in the general population. The other 50% have AD but do not have an identified genetic risk factor. Interestingly, these patients are much more likely to be insulin resistant. Although the mechanisms differ through which ApoE4 and insulin resistance initiate pathology, both promote dyslipidemia, endothelial dysfunction and damage, and oxidative stress—effects that interact with Abeta to produce the final common pathway that results in AD.
Furthermore, we now know that the CNS and the periphery are closely interrelated, and each is capable of driving AD pathology. Dietary insults provoke insulin resistance, endothelial dysfunction, and dyslipidemia—and affect the CNS. In the brain, Abeta causes insulin resistance that may drive compensatory insulin resistance in the periphery.
As Craft noted in her January 2010 Functional Medicine Update interview with Jeff Bland, PhD, over the last 10 years, a rapidly increasing number of papers have reported on co-morbid relationships in which insulin resistance appears to act as a mechanistic fulcrum, connecting hypertension, dyslipidemia, type 2 diabetes and dementia /AD.28
Insulin resistance promotes endothelial dysfunction/hypertension and dyslipidemia/atherosclerosis (and food sensitivities/allergies/gut dysbiosis – but that’s another topic necessitating its own review). Any one or any combination of these can engender a leaky blood brain barrier that impacts Abeta transport (both efflux and influx) between the brain and the periphery. The end result is increased levels of Abeta in the brain, increased Abeta-induced inflammation and damage to the endothelium, increased deposition of Abeta plaque–and AD.
The bad news is that is has become abundantly clear that no single molecular target or silver bullet will prevent or cure AD. The good news is that the latest research is moving to a model in which therapies with pleiotropic effects on metabolic function combine to decrease the risk of developing AD. The best news is that all identified risk factors for AD are modifiable. There is only one genetic risk factor, ApoE4, and actualization of its potential negative effects involves interaction with environmental factors—the incremental insults that result from poor diet, micronutrient insufficiencies and an inactive lifestyle.
Although the complexity of what has been called “the morass of intermediary metabolism” appears daunting, Dr. Crarft remains optimistic. The inter-relationships among the myriad modulators of system and cellular function offer the happy prospect that intervention at one level of the metabolic web may have beneficial effects across many levels. For example, treatment that restores insulin sensitivity also typically improves endothelial function and lipid profiles. Part II of this review will discuss what the research is revealing to be the most promising means of getting off the side roads that funnel into the Alzheimer’s Interstate.
(*Although the liver is the primary source of ketones, and a ketogenic diet increases BBB permeability to ketones from the periphery, ketones are also produced from fats within the brain by astrocytes—more on this in Part II]).25
EDITORS NOTE: We are scheduled to post part II of this article on May 14, 2010 or a bit sooner.
References
-
Borroni B, Akkawi N, Martini G, et al. Microvascular damage and platelet abnormalities in early Alzheimer’s disease. J Neurol Sci. 2002 Nov 15;203-204:189-93. ↑
Abstract -
Davì G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med. 2007 Dec 13;357(24):2482-94. ↑
Abstract -
Edwards DR, Handsley MM, Pennington CJ. The ADAM metalloproteinases. Mol Aspects Med. 2008 Oct;29(5):258-89. Epub 2008 Aug 15. ↑
Abstract -
Fahrenholz F. Alpha-secretase as a therapeutic target. Curr Alzheimer Res. 2007 Sep;4(4):412-7. ↑
Abstract -
Postina R. A closer look at alpha-secretase. Curr Alzheimer Res. 2008 Apr;5(2):179-86. ↑
Abstract -
Deuss M, Reiss K, Hartmann D. Part-time alpha-secretases: the functional biology of ADAM 9, 10 and 17. Curr Alzheimer Res. 2008 Apr;5(2):187-201. ↑
Abstract -
Stefani M, Liguri G. Cholesterol in Alzheimer’s disease: unresolved questions. Curr Alzheimer Res. 2009 Feb;6(1):15-29. ↑
Abstract -
Jaeger S, Pietrzik CU. Functional role of lipoprotein receptors in Alzheimer’s disease. Curr Alzheimer Res. 2008 Feb;5(1):15-25. ↑
Abstract -
Umeda T, Mori H, Zheng H, et al. Regulation of cholesterol efflux by amyloid beta secretion. J Neurosci Res. 2010 Feb 12. [Epub ahead of print] . ↑
Abstract -
Cossec JC, Marquer C, Panchal M, et al. Cholesterol changes in Alzheimer’s disease: Methods of analysis and impact on the formation of enlarged endosomes. Biochim Biophys Acta. 2010 Mar 26. [Epub ahead of print]. ↑
Abstract -
Jiang Y, Mullaney KA, Peterhoff CM, et al. Alzheimer’s-related endosome dysfunction in Down syndrome is Abeta-independent but requires APP and is reversed by BACE-1 inhibition. Proc Natl Acad Sci U S A. 2010 Jan 26;107(4):1630-5. ↑
Abstract -
Beel AJ, Sakakura M, Barrett PJ, et al. Direct binding of cholesterol to the amyloid precursor protein: An important interaction in lipid-Alzheimer’s disease relationships? . Biochim Biophys Acta. 2010 Mar 18. [Epub ahead of print] . ↑
Abstract -
Casserly I, Topol E. Convergence of atherosclerosis and Alzheimer’s disease: inflammation, cholesterol, and misfolded proteins. Lancet. 2004 Apr 3;363(9415):1139-46. ↑
Abstract -
Xiong H, Callaghan D, Jones A, et al. Cholesterol retention in Alzheimer’s brain is responsible for high beta- and gamma-secretase activities and Abeta production. Neurobiol Dis. 2008 Mar;29(3):422-37. ↑
Abstract -
Martins IJ, Berger T, Sharman MJ, et al. Cholesterol metabolism and transport in the pathogenesis of Alzheimer’s disease. J Neurochem. 2009 Dec;111(6):1275-308. ↑
Abstract -
Marzolo MP, Bu G. Lipoprotein receptors and cholesterol in APP trafficking and proteolytic processing, implications for Alzheimer’s disease. Semin Cell Dev Biol. 2009 Apr;20(2):191-200. ↑
Abstract -
Gasparini L, Gouras GK, Wang R, et al. Stimulation of beta-amyloid precursor protein trafficking by insulin reduces intraneuronal beta-amyloid and requires mitogen-activated protein kinase signaling. J Neurosci. 2001 Apr 15;21(8):2561-70. ↑
Abstract -
Boron W, Boulpaep E. Section III: The Nervous System in Medical Physiology, 2nd Ed. Elsevier: Philadelphia, 2009. pp. 265-408. ↑
-
Jofre-Monseny L, Minihane AM, Rimbach G. Impact of apoE genotype on oxidative stress, inflammation and disease risk. Mol Nutr Food Res. 2008 Jan;52(1):131-45. ↑
Abstract -
Ji ZS, Miranda RD, Newhouse YM, et al. Apolipoprotein E4 potentiates amyloid beta peptide-induced lysosomal leakage and apoptosis in neuronal cells. J Biol Chem. 2002 Jun 14;277(24):21821-8. ↑
Abstract -
Ji ZS, Müllendorff K, Cheng IH, et al. Reactivity of apolipoprotein E4 and amyloid beta peptide: lysosomal stability and neurodegeneration. J Biol Chem. 2006 Feb 3;281(5):2683-92. ↑
Abstract -
Jans DM, Martinet W, Van De Parre TJ, et al. Processing of amyloid precursor protein as a biochemical link between atherosclerosis and Alzheimer’s disease. Cardiovasc Hematol Disord Drug Targets. 2006 Mar;6(1):21-34. ↑
Abstract -
Herman AG. New insights into the etiopathogenesis of atherosclerosis and atherothrombosis . Bull Mem Acad R Med Belg. 2006;161(3-4):213-25; discussion 226-7. ↑
Abstract -
Clark, DD; Sokoloff L (1999). Siegel GJ, Agranoff BW, Albers RW, Fisher SK, Uhler MD. ed. Basic Neurochemistry: Molecular, Cellular and Medical Aspects. Philadelphia: Lippincott. pp. 637–70. ↑
-
Maalouf M, Rho JM, Mattson MP. The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain Res Rev. 2009 Mar;59(2):293-315. Epub 2008 Sep 25. ↑
Abstract -
Craft S. Insulin resistance syndrome and Alzheimer disease: pathophysiologic mechanisms and therapeutic implications. Alzheimer Dis Assoc Disord. 2006 Oct-Dec;20(4):298-301. ↑
Abstract -
Craft S. Insulin resistance and Alzheimer’s disease pathogenesis: potential mechanisms and implications for treatment. Curr Alzheimer Res. 2007 Apr;4(2):147-52. ↑
Abstract -
Suzanne Craft, PhD, interviewed by Jeff Bland, PhD for the January 2010 Functional Medicine Update, distributed by Synthesis. ↑
http://www.jeffreybland.com/ -
Zhao WQ, De Felice FG, Fernandez S, et al. Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J. 2008 Jan;22(1):246-60. ↑
Abstract -
De Felice FG, Vieira MN, Bomfim TR, et al. Protection of synapses against Alzheimer’s-linked toxins: insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc Natl Acad Sci U S A. 2009 Feb 10;106(6):1971-6. Epub 2009 Feb 2. ↑
Abstract -
Craft S. The role of metabolic disorders in Alzheimer disease and vascular dementia: two roads converged. Arch Neurol. 2009 Mar;66(3):300-5. ↑
Abstract
Comments are closed.