
Abstract
Accumulation of amyloid plaque is a common feature not only in Alzheimer’s disease (AD), Creutzfeldt-Jakob Disease (aka Mad Cow disease), and the dementias associated with Parkinson’s disease and Huntington’s disease, but in the aging brain, brain injury, and certain chronic low grade infections, such as Lyme disease. Amyloid-beta (Abeta or Aβ), a peptide of 39-43 amino acids, is the main constituent of amyloid plaques. While the specific mechanisms of beta-amyloid’s involvement in neuropathology remain to be fully characterized, the preponderance of evidence indicates that beta-amyloid (Abeta) accumulation plays a significant role in AD, promoting inflammation and oxidative stress, and increasing the aging brain’s vulnerability to functional impairment and neuronal atrophy.
An exponential increase in the incidence of AD, combined with the aging of the American population, brings into sharp focus an urgent need for interventions to prevent and/or remove neurological accumulation of Abeta. Unfortunately, recent reviews of once promising pharmaceutical agents indicate not only the complete failure of candidate drugs to reduce Abeta accumulation, but potentially serious side effects. Advances in diagnosis and palliative treatment of AD, and other age-related neuropathologies involving the accumulation of soluble Abeta and amyloid-beta plaque, have done nothing to lessen their rapidly escalating incidence in the United States.
This article will review current research on AD etiology, progression and available pharmaceutical options, which remain ineffective in AD prevention or treatment, and will then discuss the peer-reviewed research on promising nutraceutical agents including Turmeric Extract, Ferulic Acid, Ashwagandha Extract, Rosemary Extract, Ginkgo Biloba Extract, Red Ginseng, Myricetin, Vinpocetine, and Huperzine-A, which have demonstrated the ability to:
- reduce and eliminate soluble Abeta and Abeta plaque in the brain
- deliver potent antioxidant protection to neurons
- prevent Abeta-related neurotoxicity
- help regenerate brain neural networks
- restore Abeta-related cognitive deficits
- improve mental function in patients with AD
- improve memory and cognitive function in healthy subjects
Introduction
Alzheimer’s Disease: a 21st Century Epidemic
In a statement made in 2003, past president of the Alzheimer’s Association, Sheldon Goldberg warned, “We have 10 years, at most, to prevent disaster. If we miss that chance, Alzheimer’s will bankrupt family, state, and federal budgets as up to 14 million baby boomers succumb to the disease.” Is Goldberg an unrealistic predictor of doomsday? The most recent data suggests he may not be far off the mark.
Statistics in a March 2008 review in Biomedicine & Pharmacotherapy and in the 2008 report from the Alzheimer’s Foundation indicate1 2:
- AD currently afflicts 5.2 million or 1 in 8 individuals >65.
- Every 71 seconds, another American develops AD; by 2050, this rate will escalate to a new case of AD every 33 seconds.
Age: the Major Risk Factor for AD
- One in 5 women and 1 in 7 men who reach age 55 will develop some form of dementia in their remaining lifetime; in 1 in 6 of women >55 and 1 in 10 of men >55, the dementia will be AD.
- The number of Americans aged <65 is expected to double from approximately 35 million in 2007 to more than 75 million by 2030.3
- The number of people <65 affected by AD is projected to reach 7.7 million by 2030 – a greater than 50% increase from the 5 million affected today.
The Aging of America: a Potential Alzheimer’s Disease Epidemic
- AD accounts for 70% of all cases of dementia in Americans <71, and 90% of dementia among individuals <90. (Vascular dementia accounts for 17% of cases of dementia. Other diseases and conditions including Parkinson’s disease, Lewy body disease, frontotemporal dementia and normal pressure hydrocephalus, account for the remaining 13%.)
- It can be argued that AD is the most significant disease threat we face. From 2000 to 2005, deaths attributed to AD disease increased by 44.7%, while deaths due to heart disease, the #1 cause of death, dropped (-8.6%). Deaths due to breast cancer also dropped (-0.8%), as did deaths due to prostate cancer (-4.9%), and stroke (-14.4%). Clearly, we are making headway against heart disease and cancer, while AD has reached epidemic proportions and not only remains uncurbed, but is increasing exponentially.
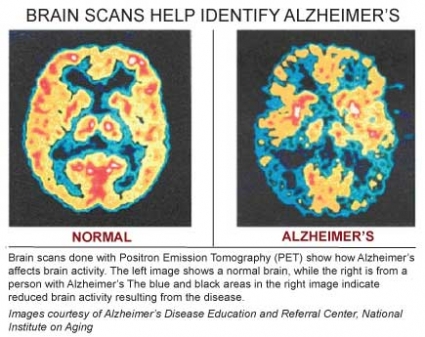
Causes of AD: the Amyloid Hypothesis5 6
In AD and other types of dementia, the rate at which nerve cells deteriorate and die rapidly increases, greatly outstripping repair and renewal mechanisms. A healthy adult brain has 100 billion neurons whose axons connect via 100 trillion synapses where neurotransmitters are released by neurons and transmit information to adjacent cells. Different strengths and patterns of chemical signals move constantly across synapses, creating the cellular basis of memories, thoughts and skills.
In AD, information transfer at the synapses in the cerebral cortex and certain subcortical regions begins to fail, the number of synapses declines, and eventually neurons die. Gross atrophy occurs in affected regions, including the temporal lobe, parietal lobe, and parts of the frontal cortex and cingulated gyrus.7 In advanced AD, the brain exhibits dramatic shrinkage from cell loss and widespread debris, not only from amyloid plaques outside and around neurons, but from neurofibrillary tangles that build up inside dying neurons.
Scientists do not yet fully understand the processes that result in the catastrophic brain damage associated with AD, but the disease has been identified as a proteopathy (protein misfolding disease) due to the accumulation of abnormally folded Abeta fibrils and tau proteins in the brains of AD patients.
The plaques seen in AD are composed of the peptide, beta-amyloid (aka Abeta or Aβ), a protein fragment snipped from a larger transmembrane protein called amyloid precursor protein (APP). APP is located in the neuron’s membrane where, in the non-amyloidogenic pathway, it is cleaved by the a-secretase enzyme, producing “a-secretase cleaved soluble APP (APPsa), which is thought to play a role in neuronal survival and repair after injury. In the amyloidogenic pathway, however, APP is proteolytically processed by the beta- and gamma-secretases to release the neurotoxic Abeta peptide.
Abeta peptide first undergoes conformational changes into oligomers or protofibrils, which then assemble into highly ordered beta-sheet structures referred to as amyloid fibrils. These then self-aggregate into the misfolded proteins that deposit as the hallmark amyloid plaques seen in the brains of patients suffering from AD.8
The neurofibrillary tangles, seen within the neurons in AD, are the result of the aggregation of abnormally folded tau protein. In healthy neurons, tau protein phosphorylates, and thus stabilizes, the microtubles that constitute the internal structure of the neuron and provide the pathway through which nutrients and other molecules move from its body out to the ends of the axon and back. In AD, tau becomes hyperphosphorylated, and threads of hyperphosphyorylated tau join, forming the characteristic neurofibrillary tangles seen inside affected nerve cell bodies.
Of the three major competing hypotheses to explain the central mechanism in AD, the most evidence supports Abeta as the primary causative agent. Not only is the cytotoxicity of mature aggregated amyloid fibrils, which disrupt the neuron’s calcium ion homeostasis and thus induce apoptosis, well recognized, but the latest research indicates that soluble Abeta (Abeta in oligomer form) is even more toxic than its final aggregated plaque form.
The other two hypotheses are the cholinergic hypothesis and the tau hypothesis. The cholinergic hypothesis, the basis for most currently available drug therapies, suggests the central problem is reduced biosynthesis of the neurotransmitter acetylcholine. However, this appears unlikely as medications that treat acetylcholine deficiency only palliate symptoms briefly (3-6 months), produce heptatotoxicity, and do nothing to halt or reverse AD.9
The tau hypothesis differs from the beta-amyloid hypothesis only in suggesting that tau protein abnormalities initiate the disease cascade since these have a stronger correlation with neuron loss than amyloid plaque deposition. However, a growing body of genetic and biochemical evidence suggests the formation of neurofibrillary tangles is triggered by abnormal Abeta accumulation, and soluble Abeta has been shown to induce abnormal tau phosphorylation.10 11
The amyloid hypothesis is further supported by the fact that the gene for APP is located on chromosome 21, and patients with Downs Syndrome, who have trisomy 21 (and thus an extra copy of the APP gene), almost universally exhibit AD-like disorders by age 40.
Another piece of data supporting the Abeta hypothesis is the fact that ApoE4, one genetic variant of the apoproteins that transport cholesterol, is known to be a major risk factor for AD as well as cardiovascular disease. (The ApoE4 isoform is associated with increased plasma LDL levels, putting carriers at increased risk for atherosclerosis and coronary heart disease.) ApoE4, a major risk factor present in more than 40% of patients with dementia, also binds to APP, promoting Abeta production and fulminant amyloid deposition in the brain.12
Trouble begins when ApoE4 and other yet-to-be-identified factors trigger overproduction of Abeta or reduce the brain’s ability to dispose of it. The excess jams signaling at the synapses, blocking information flow and leading to a cascade of damaging events ending in cell death.13 14
Amyloid-beta Oligomers: a Modification of the Amyloid Plaque Hypothesis
The lack of correlation between amyloid plaque number and severity of dementia has frequently been cited as evidence against the hypothesis that amyloid plaque is the key player in AD. A correlation has, however, been shown between soluble Abeta levels and the extent of synaptic loss/severity of cognitive impairment, leading to the suggestion that oligomers of Abeta, the building blocks of insoluble amyloid plaques, are the earliest mediators of neuronal dysfunction. Data have accumulated indicating that soluble non-fibrillar forms of Abeta are proximate effectors of synaptotoxicity. In vitro research has shown a dramatic decrease in the number of dendritic spines when neurons were exposed to sub-nanomolar concentration of Abeta oligomers. Furthermore, the spine loss was reversible upon addition of anti-Abeta monoclonal antibodies. Shah RS et al., among other researchers in the field, argue that “amyloid-beta plaques may be more pathognomonic than pathogeneic.”15 16
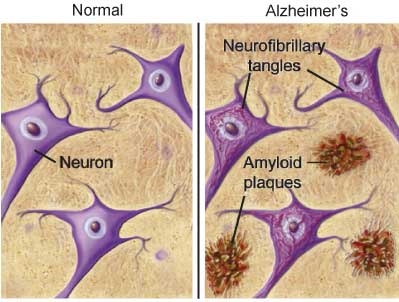
Amyloid Plaques
{kind=link}
Amyloid plaques, and neurofibrillary tangles—visible, respectively, between and inside affected neurons—are the hallmarks of AD. Both not only disrupt neuronal communication and reduce the neuron’s ability to resist or recover from injury, but promote inflammation and oxidative damage, further contributing to neurons’ degradation and resulting AD symptomatology.
Young versus Senile AD
Although onset of AD is usually seen in persons >65 (referred to in the medical literature as “senile AD”), the condition presents in individuals as young as 30. In young AD, (AD in persons >50) a rare condition accounting for only ~2% of all cases, a strong correlation has been noted between dementia severity and greatly increased amyloid plaque and neurofibrillary tangle burden, whereas this association has not been found in older patients with the disease. In senile AD, cerebrovascular pathology findings, typically indicative of age-related degeneration, are more severe. For this reason, it has been suggested that young AD is more likely to involve increased genetic susceptibility (e.g., ApoE4), while senile AD is more likely to be due to a combination of genetic and environmental factors, including age-related and cerebrovascular changes as well as AD pathology.17 18
Pharmaceutical Agents Ineffective
The search for pharmaceutical options to prevent and treat AD has spurred research into potential amyloid inhibitors. Beta-secretase processing of amyloid precursor protein (APP), the first step in the pathway leading to the production of Abeta, has been a major target; however, development of beta-secretase inhibitors has been slow. Candidate molecules not only adhered to themselves, forming aggregates unable to cross the blood-brain barrier, but lost their specificity, binding not only to amyloid but any available protein, inhibiting protein chemistry in an uncontrolled manner.19 Recent progress has evolved inhibitors in sizes sufficiently small to penetrate cell membranes and the blood-brain barrier, yet retain potency for the inhibition of Abeta production in cultured cells and experimental animals; however, even if clinically useful Abeta inhibitors can eventually be developed, such agents remain years away from availability.20
gamma-Secretase, a multi-protein complex that proteolyzes the transmembrane region of APP, producing Abeta, has also been a top drug target. Unfortunately, this protease cleaves not only Abeta, but a number of other substrates, including the Notch receptor. Their potential for interference with the Notch signaling pathway, which appears to play a major role in regulating cell fate decisions, renders gamma-secretase inhibitors highly problematic.21 The most fully developed AD-modifying pharmaceutical agent, tramiprosate, is a sulfated glycosaminoglycan mimetic that binds to soluble Abeta, preventing aggregation. However, the only human trial so far conducted, a small double-blind study (n=58) of subjects with mild to moderate AD, showed no improvement on cognitive or clinical measures after 3 months of treatment.22 In addition, other research indicates that tramiprosate promotes abnormal aggregation of tau, the other histopathological hallmark of AD.23
Other drugs currently used to slow AD progression include statins, NSAIDs and memantine. Statins slow progression slightly but are unable to reverse neuronal degeneration once it has occurred. Non-steroidal anti-inflammatory drugs (NSAIDs) downregulate pro-inflammatory signals, microglia, and astrocytes, and may reduce risk of AD by lowering Abeta production. However, randomized controlled trials of various NSAIDs have failed to show a significant effect on the rate of cognitive decline in patients with dementia or the rate of conversion to AD in patients with mild cognitive impairment. Moreover, a randomized controlled primary prevention trial of NSAIDs in AD, the AD Anti-inflammatory Prevention Trial (ADAPT), was terminated in 2004 due to concerns regarding cardiovascular risk.
Memantine helps prevent excitotoxicity by binding to the NMDA glutamate receptor with a higher affinity than Mg2+ ions, thus inhibiting the prolonged influx of Ca2+ ions that causes neuronal excitotoxicity. On the plus side, memantine’s low affinity and fast off/on kinetics let it sit on the receptor just long enough to prevent pathologic activation of the glutatmate receptors, but not so long that it interferes with normal NMDA activation. When physiologic activation of NMDA receptors is required for learning and memory, they can still be activated by the relatively high concentrations of glutamate released following depolarization of the presynaptic neuron. However, although NMDA-receptor-dependent excitotoxicity contributes to the progressive neuronal loss seen in AD, the weak NMDA-receptor blocking property of memantine has produced only minimal symptomatic improvement in AD patients.24
In sum, available pharmacetucial options are neither effectively addressing AD symptoms nor preventing its progression.25
Nutraceuticals with Significant Therapeutic Potential
A growing body of research indicates that a number of nutraceuticals, including Curcumin, Rosemary Extract, Ginkgo Biloba Extract, Red Ginseng, Vinpocetine, Huperzine-A, Ferulic Acid, Myricetin, and Ashwagandha Extract, show significant promise in:
- preventing amyloid plaque formation
- reducing and removing soluble Abeta and beta-amyloid plaques
- supporting regeneration of brain neural networks
- providing neurological antioxidant protection
- enhancing cognition in healthy individuals
- improving mental function in patients with AD
These nutraceuticals are available now, offer remarkably few, if any, side effects, and taken together, may greatly aid in managing brain senescence.
Current Research on Promising Nutraceuticals
1. Curcumin
Curcumin is the most active of the three curcuminoids found in the yellow-orange Indian curry spice, turmeric (Curcuma longa); the other two are demethoxycurcumin and bis-demethoxycurcumin.
In vitro research has shown curcumin to be a pleiotropic anti-amyloid, antioxidant, anti-inflammatory and immune-modulating treatment for AD.26
Anti-amyloid: By directly binding small Abeta species, curcumin blocks Abeta
aggregation and fibril formation in vitro and in vivo. Adding curcumin to pre-aggregated oligomers results in the appearance of intense monomeric bands, indicating de-aggregation.27
Anti-oxidant: A number of in vitro studies, including research presented at the American Physiological Society’s 2004 annual conference in Washington, D.C., have confirmed that curcumin strongly induces expression of the gene hemeoxygenase-1 (HO-1) in astrocytes from the hippocampal region of the brain. HO induction, by generating the vasoactive molecule carbon monoxide and the powerful antioxidant bilirubin, is a protective system against brain oxidative injury. Curcumin also exerts potent antioxidant activity against NO-related radical generation.27 28
Anti-inflammatory: Most inflammatory stimuli activate 1 of 3 independent MAPK pathways, which lead to activation of the p44/42 MAPK (also called ERK1/ERK2), JNK, or the p38 MAPK pathway, respectively. Curcumin inhibits all three pathways directly or indirectly.29 27
Metal chelation: A consistent observation in AD is a dysregulation of metal ions [(Fe(2+), Cu(2+) and Zn(2+)] homeostasis and consequential induction of oxidative stress, associated with Abeta aggregation and amyloid plaque formation. In addition, Abeta has been shown to spontaneously self-aggregate in the presence of divalent metals (Fe2+, Cu2+, Zn2+) into neurotoxic amyloid fibrils in the neocortex. Metal chelation is known to reduce both oxidative stress and amyloid plaque formation. Curcumin has well characterized chelating actions and readily binds both Cu2+ and Fe2+.30 27
Immune modulation: Over the last two decades, curcumin has been shown to be a potent immunomodulatory agent that can regulate the activation of T cells, B cells, macrophages, neutrophils, natural killer cells, and dendritic cells. Curcumin can also downregulate the expression of various pro-inflammatory cytokines including TNF, IL-1, IL-2, IL-6, IL-8, IL-12, and chemokines, most likely through inactivation of the transcription factor NF-kappaB. Interestingly, however, at low doses, curcumin also enhances antibody responses.31 32
Curcumin in Animal Models of AD
In numerous animal models of AD, curcumin has been shown to cross the blood-brain barrier and reduce senile plaques and cerebrovascular amyloid angiopathy.33
Systemic treatment of mice with curcumin for 7 days clears existing plaques, indicating a potent disaggregation effect. Curcumin also causes significant reversal of structural changes in dystrophic dendrites, including abnormal curvature and dystrophy size. Together, these data suggest that curcumin reverses existing Abeta pathology and associated neurotoxicity in a mouse model of AD.34
Animal studies have demonstrated that curcumin not only clears Abeta from the brain, but reduces AD-related inflammation in brain tissue. Curcumin’s brain antioxidant effects have been well-documented in animal models of cerebral ischemia and brain trauma. Not only has plaque buildup been reduced in curcumin-fed rats, but they also outperformed rats on control diets when carrying out maze-based memory tests.27
In AD, several mediators in the inflammation cascade contribute both to neurodegeneration and the production and accumulation of Abeta peptide. Animal studies have shown that curcumin antagonizes many steps in the inflammatory cascade, including AP-1 transcription, activation of NF-kappaB, iNOS and JNK.27
IL-1, an index of neuroinflammation, is also thought to contribute to AD pathogenesis and is elevated in animal models of AD. IL-1 is effectively reduced in curcumin-fed mice compared to controls. The stress-activated (phosphorylated) c-Jun N-terminal kinase (pJNK), also elevated in animal models of AD, is significantly reduced by curcumin as well, and the degree of soluble Abeta reduction is proportional to that of pJNK reduction.27 (JNK is a tau kinase hypothesized to mediate end stage neurodegeneration in AD models.)
The consensus of researchers conducting animal studies has been unanimous: curcumin’s extremely low incidence of side effects, combined with its ability to reduce Abeta burden, and anti-inflammatory and anti-oxidant actions, indicate significant therapeutic potential in combating AD in humans.35 36 37
“The prospect of finding a safe and effective new approach to both prevention and treatment of Alzheimer’s disease is tremendously exciting,” noted Gregory Cole, MD, a professor of medicine and neurology at the David Geffen School of Medicine at UCLA, in a news release after UCLA research showed curcumin not only inhibits Abeta aggregation and dissolves amyloid fibrils, but does so more effectively than ibuprofen and naproxen. Previous studies had shown that people taking these NSAIDs have a decreased risk of developing Alzheimer’s disease.38 34
Curcumin in Human Research Epidemiological studies have noted very low levels of AD and other dementias in elderly Indian populations whose typical diet is liberally spiced with turmeric. In fact, studies have found that AD affects just 1% of people over the age of 65 living in some Indian villages.39
Similar findings have been noted in other populations. In a recent population-based study of 1,010 elderly non-demented Asians, those who consumed curry ‘‘occasionally’’ and ‘‘often or very often’’ scored significantly better on the Mini-Mental State Examination (MMSE), an established measure of cognitive function, than did those who “never or rarely” consumed curry.40
Bisdemethoxycurcumin restores innate immunity & Abeta clearance in AD patients
Phagocytosis of Abeta by macrophages is excellent in normal subjects but deficient in most AD patients. Increased proinflammatory cytokine levels and activated microglia and macrophages in AD patients may be compensatory for defective clearance of Abeta. Consequently, therapeutic interventions that increase phagocytosis of Abeta might decrease brain inflammation as well as reduce inflammation-induced neurodegeneration.
A secondary curcuminoid in Curcuma longa, bisdemethoxycurcumin, has been shown to improve innate immune system activity in AD patients, increasing Abeta clearance from the brain. In healthy individuals, Abeta presentation stimulates upregulation of genes that increase transcription of beta-1,4-mannosyl-glycoprotein
4-beta-N-acetylglucosaminyltransferase (MGAT3) and other genes, including Toll-like receptors. These increase macrophage transport of Abeta into endosomes and lysosomes; in AD patients, however, an increase in Abeta generally down-regulates these genes, resulting in defective phagocytosis. Using blood samples from AD patients, Drs. Milan Fiala and John Cashman have shown that bisdemethoxycurcumin enhances transcription of MGAT3 and Toll-like receptors, restoring macrophage activity to normal levels and enhancing Abeta phagocytosis.41 42
Curcumin extremely safe and well tolerated
At least three different Phase I clinical trials have indicated that curcumin is extremely safe and well tolerated when taken in doses as high as 12 g/day. These results were further confirmed in a dose-escalation trial to determine curcumin’s maximum tolerated dose and safety in which a standardized powder extract of curcumin was administered to 24 healthy volunteers in single doses ranging from 500 to 12,000 mg. Only minimal, non-dose-related toxicity was seen and only in seven subjects (30%).43
Bioavailability enhanced by piperine, the active phytochemical in black pepper
It has been noted that curcumin is extremely rapidly metabolized via glucuronidation in the liver and intestinal wall, which some believe may limit its therapeutic usefulness. Piperine, a known inhibitor of glucuronidation, has been shown to significantly enhance curcumin’s bioavailability in studies involving both rats and healthy human volunteers. In rat studies, administration of curcumin alone at a dose of 2 g/kg, resulted in only moderate serum concentrations over 4 hours. Concomitant administration with piperine (20 mg/kg) increased the serum concentration of curcumin for 1-2 hours post drug, significantly increased the time to maximum concentration, and significantly decreased elimination half-life and clearance, increasing bioavailability by 154%. In humans, administration of curcumin alone produced undetectable or trace amounts in serum; however, concomitant administration with piperine (20 mg/kg) resulted in much higher concentrations and increased bioavailability by a remarkable 2000%.44
Interestingly, black pepper is a common ingredient, along with curcumin, in curry spice blends, which may contribute to the beneficial effects of turmeric consumption seen in epidemiological studies in India and other areas where curries are standard fare.
2. Rosemary Extract
Rosemary has a long history of folk use as a brain stimulant, remedy for heart trouble, antiseptic, insect repellent, and food preservative. Rosmarinic acid, a polyphenol antioxidant carboxylic acid found in rosemary (and other Lamiaceae herbs commonly used as culinary herbs, including oregano, sage, thyme and peppermint) is metabolized in both rats and humans to methylated rosmarinic acid, coumaric acid, ferulic acid and caffeic acid.
Recent research has shown that rosmarinic acid, in addition to being a potent antioxidant, is also antiviral, antibacterial, anti-inflammatory, and protects brain cells from Abeta toxicity.45
An in vitro study at the University of Naples, Italy, demonstrated the neuroprotective effect of rosmarinic acid on rat neurons exposed to the toxic effects of Abeta. Rosmarinic acid was found to reduce reactive oxygen species (ROS) formation in addition to lipid peroxidation, DNA fragmentation, and tau hyperphosphorylation—all key factors in AD. The researchers concluded that their results validated the use of rosemarinic acid in patients suffering from mild to moderate AD because “this natural compound exerts neuroprotective, antioxidative, and anti-apoptotic effects against Abeta insult.”46
In addition to preventing the toxicity of Abeta to brain cells, rosmarinic acid has also been found to inhibit Abeta formation (most likely due to its inhibition of beta-secretase) and destabilize and dissolve Abeta fibrils that have already formed. The results were so impressive, the researchers concluded that rosmarinic acid “could be a key molecule for the development of therapeutics for Alzheimer’s disease.”47 45
Another recent in vivo study also showed that rosmarinic acid exerts an anxiolytic effect while supporting short- and long-term memory retention.48
3. Ginkgo Biloba Extract
Ginkgo biloba extract has demonstrated neuroprotective effects in a variety of animal models and has improved or maintained cognitive function in AD patients.
Ginkgo biloba exerts vaso-regulatory actions that protect blood vessels and help explain its effectiveness in managing cerebral insufficiency states, neurosensory disturbances and peripheral occlusive arterial disease. Additionally, Ginkgo biloba extract has been shown to abolish Abeta-induced ROS generation and to protect, in a dose-dependent manner, against Abeta-induced apoptosis and neurotoxicity.
Ginkgo biloba extract also inhibits, in a dose-dependent manner, the formation of Abeta-derived soluble neurotoxic oligomers. These diffusible, nonfibrillar ligands derived from Abeta are potent CNS neurotoxins, which assemble when fibril formation is inhibited, and kill neurons at nonomolar concentrations. Soluble ligands of Abeta have been found in AD brains, where their levels were 12-fold higher than those present in normal brains.49
A study published in the May 2004 issue of the Journal of Neuroinflammation reported that brain cells pre-treated with nanomolar concentrations of Ginkgo biloba extract before exposure to beta-amyloid protein were resistant to its toxic effects and survived without damage.50
In male Wistar rats subjected to 20 minutes of ischemia followed by 7 days reperfusion, 7 days pretreatment with Ginkgo biloba (40 mg/kg/day) significantly decreased oxidative damage in the forebrain, protecting neurons in the vulnerable CA1 pyramidal layer of the hippocampus; 20 minutes of ischemia is usually lethal for these cells.51
In vitro research conducted at Georgetown University Medical Center, Washington, D.C., confirmed that nerve cells exposed to Abeta experience increased ROS production and cellular death. If pre-treated with Ginkgo biloba extract, however, ROS production, cellular damage and death were all inhibited.52 Likewise, a study at the University of Southern Mississippi found that brain cells treated with Ginkgo biloba extract were resistant to the damaging effects of Abeta.53
In vivo research conducted at Georgetown University Medical Center suggests that the mechanism through which Gingko biloba inhibits Abeta formation and rescues neuronal cells from Abeta-induced cell death involves its ability to decrease the capacity of low density lipoproteins (LDL) to carry free cholesterol to various tissues without affecting the capacity of high-density lipoproteins (HDL) to carry cholesterol back to the liver.
High cholesterol levels affect APP generation and Abeta processing since one of the physiological functions of APP and Abeta is to control free cholesterol transport. High levels of free cholesterol induce over-production of APP mRNA expression and Abeta. Specifically, in the presence of high cholesterol levels, APP synthesis (expression) is increased, and the processing of APP for the formation of Abeta and Abeta peptide fragments is enhanced. In addition, Abeta increases the capacity of lipoprotein and apolipoprotein to carry free cholesterol, which may explain the link between the ApoE4 genotype and increased risk for cardiovascular disease as well as AD.
In additional research conducted at Georgetown, long-term treatment of aging Brown Norway rats with Gingko biloba extract lowered production of brain APP and Abeta by decreasing the capacity of LDL to carry free cholesterol, thus lowering levels of circulating free cholesterol. These in vivo results indicate a mechanism through which Gingko biloba extract inhibits the cerebral amyloidogenesis and Abeta angiopathy that characterize all forms of AD.54
4. Red Ginseng Extract
Also called Panax ginseng, Red ginseng is one of the most heavily researched adaptogens and the most commonly used Asian ginseng. Thousands of studies have demonstrated that Ginseng provides a myriad of beneficial effects ranging from maintaining normal glucose levels to stimulating immune function. Red ginseng extract has also been found to enhance cognitive function. Two ginseng phytochemicals in particular—ginsenosides Rb1 and Rg1—have demonstrated the ability to improve memory in animal models of AD.55
Recent Japanese studies showed that in addition to improving memory, the ginseng extracts were able to regenerate brain axons and synapses in laboratory animals (in vivo and in vitro), a highly significant finding as these neurons are typically destroyed in AD.55 56
Ginsenoside Rg2 has been shown to protect neurons against neuronal injury and Abeta formation induced by glutamate. Although glutamate is a principal excitatory neurotransmitter in the brain and is involved in many neurological functions, including cognition and memory, excessive activation of glutamate receptors can cause neuronal injury or death. Such glutamate-related injury is induced by excessive calcium influx into neurons triggered by activation of glutamate receptors.
In addition to its direct excitotoxic effects on neurons, glutamate can also induce formation of Abeta by activating caspase-3, which cleaves APP during apoptosis. Rg2 inhibits excessive Ca2+ influx triggered by glutamate, reduces lipid peroxidation and down-regulates the expression of caspase-3.57
An in vitro study performed at the College of Pharmacy, Chung-Ang University, Seoul, Korea, found that Red ginseng extract was able, via its immune-stimulating effects, to reduce inflammation in neurons exposed to Abeta by promoting its microglial phagocytosis.58
Studies using the mouse model of AD have indicated that even a single, orally administered dose of ginsenoside Re, Rg1, or Rg3, results in a significant reduction the amount of Abeta detected in the animals’ brains 18 hours postdrug administration. Importantly, these effects were observed after an administration of only 25 mg/kg of the gionsenosides.59
In vitro research conducted at the University of Alberta, Canada, demonstrated that ginseng saponins can reverse the inhibitory effect of Abeta on acetylcholine release in hippocampal neurons.60 When given orally to rats (5 days before and 7 days after Abeta injection), ginseng saponins (80 mg/kg/day) reversed cognitive impairment, possibly by minimizing Abeta’s inhibitory effect on hippocampal cholinergic transmission. However, treating the animals with ginseng only after Abeta injection was ineffective in obliterating Abeta’s amnesiac effect.61
The combined results of these in vitro and animal studies strongly indicate that ginseng itself, or purified ginsenosides, may have similarly useful effects in human AD. And also suggest prophylactic use of ginseng may be protective against AD.
5. Vinpocetine
Derived from the periwinkle plant Vinca minor, vinpocetine is also showing a number of neuroprotective properties that may help in the prevention and treatment of AD and other neurodegenerative pathologies involving oxidative stress. Vinpocetine is an antioxidant and vasodilator, increasing blood flow to the brain.
In a series of in vitro studies, researchers from the University of Coimbra in Portugal have shown that vinpocetine blocks inhibition of the mitochondrial respiratory chain complexes II,III and IV, completely abolishes the depletion of pyruvate levels induced by toxic concentrations of Abeta peptides, and prevents the generation of oxidative stress due to excessive accumulation of ROS in cells treated with Abeta.62
6. Huperzine A
Huperzine A, a Lycopodium alkaloid extracted from the Chinese moss, Huperzia serrata, is a potent, reversible, selective inhibitor of acetylcholinesterase (AChE), and has a long history of effective, safe use in Traditional Chinese Medicine. The potency of Huperzine A in AChE inhibition is similar or superior to that of galanthamine, donepezil, rivastigmine and tacrine, and it has been approved as a drug in China to treat AD and other age-associated memory impairments. Clinical trials in China have demonstrated that Huperzine A significantly relieves memory deficits in the elderly related to AD and vascular dementia, without the side effects that usually accompany pharmaceutical acetylcholinesterase inhibitors.63 64
Although the complex nature of AD pathology is well recognized, available drug therapies target only single components of cognition, which may be one reason they demonstrate only palliative effect. Huperzine A has shown itself to be a multifunctional agent with effects not only on AChE, but APP processing, nerve growth factor (NGF), and Abeta neurotoxicity—all of which strongly suggest the potential usefulness of this, and other nutraceuticals with multiple effects, in the treatment of AD.65 66
In vitro, animal models and human clinical trials all indicate that Huperzine A possesses the ability to not only inhibit AChE, but to:65 66
Protect nonamyloidogenic APP processing. Two pathways for APP processing have been described: the nonamyloidogenic pathway releases secretory amyloid precursor protein a (sAPPa), which prevents Abeta formation, whereas the amyloidogenic pathway produces Abeta. APP produced by the nonamyloidogenic pathway also has potent neurotrophic and neuroprotective activities against excitotoxic and oxidative insults, promotes neurite outgrowth, regulates synaptogenesis, exerts trophic effects on cerebral neurons in culture, and stabilizes neuronal calcium homeostasis.
In in vitro studies, huperzine A has been shown to increase sAPPa levels and decrease Abeta levels. Huperzine A improves APP non-amyloidogenic processing by increasing the levels of protein kinase C (PKC) isoform a, which activates various neurotransmitter receptors, including muscarinic acetylcholine receptor subtypes M1 and M3, which are thought to increase the release of sAPPa and decrease Abeta levels. PKC is also thought to regulate downstream signaling pathways, including mitogen-activated protein kinase (MAPK) signaling, which is also involved in the APP non-amyloidogenic pathway, although the precise mechanism still needs clarification.
Protect against neuronal apoptosis induced by Abeta, hydrogen peroxide, oxygen-glucose deprivation, serum deprivation, and the PKC inhibitor staurosporine.
Protect neurons against Abeta-induced oxidative stress. Abeta interaction with neuronal membranes induces the formation of peroxides and lipid peroxidation. In the early stages of AD, patients are more likely to exhibit increased oxidative stress than apoptosis. Rat neurons pre-treated with huperzine for just 2 hours before being exposed to a solution containing Abeta showed significantly less cellular stress than untreated cells, which experienced a rapid decline in ATP production. As a result, mitochondrial membrane homeostasis and integrity were disrupted, producing severe oxidative stress and death. Incubation of cortical neurons with Huperzine A, however, resulted in markedly less oxidative stress and death. In in vivo research, Huperzine A increased the antioxidant enzymes, glutathione peroxidase and catalase, decreased the level of the lipid peroxidation product malondialdehyde, and significantly reduced the rise in concentrations of superoxide dismutase and malondialdehyde in the hippocampus, cerebral cortex and serum of aged rats.
Improve nerve growth factor (NGF) signaling. In vitro experiments have shown that decreased supply of NGF to basal forebrain acetylcholine-containing neurons leads to neuronal shrinkage mimicking that seen in AD. NGF cannot be given directly as it cannot penetrate the blood-brain barrier. Incubation of neurons with Huperzine A increases NGF mRNA and protein levels, and in a rat model of transient cerebral ischemia and reperfusion-induced cognitive deficiency, resulted in improved learning and memory.
Improve neurotransmission. The central nervous system is a complex network in which one neurotransmission system often affects others, e.g., acetylcholine, noradrenaline and dopamine systems interact, influencing learning and memory. Abnormalities of all these neurotransmitters are seen in AD. Huperzine A administration markedly restores not only acetylcholine, but noradrenaline and dopamine levels. Huperzine A also acts as an NMDA antagonist (the effect of memantine, which combined with an AChE inhibitor, donepezil, has been helpful in AD).
Reverse effects of amnesia. Scopolamine, one of a class of amnesiac drugs used when cardiovascular patients undergo an angiogram and angioplasty, causes short-term memory loss. A study performed on young and old monkeys given the drug showed that Huperzine A reversed scopolamine-induced memory and performance deficits.67
Improve memory and learning in adolescents. Chinese researchers selected 34 matched pairs of healthy middle school students whose only complaints were poor memory and difficulty learning. In a double-blind trial, one member of each pair was given 100 mcg of Huperzine A twice daily for 4 weeks, while the other member received placebo. Students’ memory quotients and academic performance in Chinese, English and mathematics were measured before and after the trial. At the end of the trial, the Huperzine A group scored significantly higher than the control group on standard memory tests and also did significantly better in their Chinese and English lessons, but not in math. No side effects of any kind were noted.68
7. Myricetin
A flavonoid commonly found in foods, such as berries, vegetables, teas, wine, and herbs, myricetin has been shown to have antioxidant and anti-inflammatory properties, and to protect neurons from Abeta-associated neurotoxicity and inhibit Abeta accumulation.
Accumulation of the excitatory neurotransmitter glutamate is involved in the etiology of many neurodegenerative disorders including AD. Glutamate-mediated excitotoxicity is caused by intracellular Ca(2+) overload via the N-methyl-D-aspartate receptor (NMDA receptor), reactive oxygen species (ROS) generation, and caspase-3 activation—each of which are inhibited by myricetin. Myricetin modulates the NMDA receptor by phosphorylation, reducing glutatmate-induced Ca (2+) overload; inhibits ROS production caused by glutatmate; and reduces glutatmate-induced activation of pro-apoptotic capsase-3.69
In addition, myricetin activates and upregulates alpha-secretase and directly binds to and inhibits beta-secretase. Myricetin’s interference with these two enzymes results not only in a significant decrease in Abeta formation, but the dissolution of previously formed Abeta deposits.70
8. Ferulic Acid
An abundant phenolic phytochemical with anti-inflammatory and antioxidant properties, ferulic acid is found in the seeds of plants such as rice, wheat and oats, as well as in coffee, apple, artichoke, peanut, orange and pineapple. The chemical structure of ferulic acid closely resembles that of curcumin71, and like curcumin, ferulic acid is not only a powerful antioxidant, but a strong inducer of the heat shock response. Heat shock proteins are a highly conserved CNS mechanism responsible for the preservation and repair of correct protein conformation. Not only AD, but Parkinson’s disease, Huntington’s disease, Amyotrophic lateral sclerosis and Friedreich ataxia are all considered misfolded protein pathologies.
In vivo research has demonstrated ferulic acid’s ability to protect neurons against Abeta-induced oxidative stress and neurotoxicity, while in vitro studies have shown that ferulic acid not only inhibits Abeta formation, but destabilizes preformed Abeta.72 73
Oxidative stress is a key mechanism of Abeta-induced neurotoxicity. In a study conducted at Meijo University, Japan, pretreatment of mice with ferulic acid (5 mg/kg), once daily for 6 days, inhibited the induction of short-term memory deficits caused by an inhibitor of glutathione synthesis (buthionine-sulfoximine).74
In animal models of brain dysfunction, pre-treatment with ferulic acid protects laboratory animals injected with Abeta from developing learning and memory deficits. In a study at the Institute of Natural Medicine, Hallym University, Chunchon, South Korea, mice were allowed free access to drinking water containing ferulic acid for 4 weeks before being injected with Abeta. While control mice exhibited impaired performance on memory and behavior tests, no decrease in performance skills was seen in mice treated with ferulic acid.75
9. Ashwagandha (root of Withania somnifera)
Ashwagandha holds a place in Ayurvedic pharmacology similar to ginseng in Chinese medicine as a rejuvenating herb and powerful adaptogen that helps increase energy, endurance, and libido, while strengthening the immune system. Withanolides, the active constituents in ashwagandha, are thought to be responsible for its multiple medicinal applications in India, where this botanical is used to treat mental deficits, including amnesia, in geriatric patients. In addition to their anti-inflammatory, anti-stress, and rejuvenating properties, withanolides have recently been shown to regenerate vital neural connections and restore cognitive function in animal models of AD.76
Two Japanese studies have shown that ashwagandha stimulates the growth of axons and dendrites in human neuroblastoma cells and rat neurons, suggesting this botanical might help compensate for and repair damaged neuronal circuits in the aging brain. In further studies, in both cultured rat neurons damaged by Abeta and mice severely cognitively-impaired due to Abeta injection, withanolide A (one of the major active constituents in Ashwagandha) caused remarkable axonal and dendritic regeneration and synaptic reconstruction, restoring neural networks in vitro and Abeta-induced memory deficits in vivo.76 77 78
In numerous additional recent studies at the Institute of Natural Medicine, Toyama Medical and Pharmaceutical University, Sugitani, Japan, the same Japanese researchers found that ashwagandha’s withanolides produced significant improvement in the memory deficits of Abeta-injected rats. Withanolides not only prevented a loss of axons, dendrites and synapses, but regenerated neurites and reconstructed synapses in severely damaged neurons in memory-deficient mice showing neuronal atrophy and synaptic loss.{ref79,{ref80,81
When German researchers from the University of Leipzig injected mice with withanolides and examined slices of the animals’ brains, they found an increase in acetylcholine receptors and cholinergic activity, one mechanism that may help explain the cognition-enhancing and memory-improving effects observed.82
Conclusion
As the population ages, a dramatic increase is projected in AD. Currently, pharmaceutical treatments for AD offer only short-term palliative relief. It is clear that Abeta oligomers are deleterious to neurons in their vicinity, and emerging therapies are likely to be based on preventing such assemblies from forming and/or persisting.
A number of nutraceutical agents, including Curcumin, Rosemary Extract, Ginkgo Biloba Extract, Red Ginseng, Vinpocetine, Huperzine-A, Ferulic Acid, Myricetin, and Ashwagandha Extract, have demonstrated effective modulation of key mechanisms underlying Abeta pathology with virtually no side effects, except desirable ones. Numerous peer-reviewed studies suggest these agents can prevent and even reverse accumulation of Abeta oligomers and plaque formation.
Although these nutraceutical agents lack large scale human trials proving effectiveness in AD, their use in clinical practice is strongly supported by: their demonstrated effects on the underlying mechanisms of AD pathology, their lack of adverse effects, their other known health benefits, and the lack of any better therapeutic choices at this time. Particularly for individuals at high risk, e.g. those with an ApoE4 genotype, preventive action is imperative as optimal outcomes are clearly dependent upon prophylactic treatment.
References
-
Shah RS, Lee HG, Xiongwei Z, et al. Biomed Pharmacother. 2008 Mar 17 [Epub ahead of print]. ↑
-
Plassman BL, Langa KM, et al. Neuroepidemiology. 2007;29:125-32. ↑
-
Above image from. ↑
http://www.living-with-alzheimers-disease.com/Image1.jpg -
Irvine GB, El-Agnaf OM, Shankar GM, Walsh DM. Protein aggregation in the brain – the molecular basis for Alzheimer’s and Parkinson’s Diseases. Mol Med. 2008 Mar 27 [Epub ahead of print]. ↑
-
Shah RS, Lee HG, Xiongwei Z, et al. Current approaches in the treatment of Alzheimer’s disease. Biomed Pharmacother. 2008 Mar 17 [Epub ahead of print]. ↑
-
Wenk GL. J Clin Psychiatry. 2003;64 Suppl 9:7-10. ↑
-
Lashuel HA. “Protein fibrillogenesis in neurodenerative diseases from biophysics to therapeutic strategies”. ↑
http://nmnf.epfl.ch/page9107.html -
Shah RS, Lee HG, et al. ↑
-
Schmitz C, Rutten BP, Pielen A, et al. Am J Pathol. 2004 Apr;164(4):1495-502. ↑
-
Irvine GB, El-Agnaf OM, Shankar GM, et al. Mol Med. 2008 Mar 17 [Epub ahead of print]. ↑
-
Hass S, Fresser F, Köchl S, et al. Physical interaction of ApoE with amyloid precursor protein independent of the amyloid Abeta region in vitro. J Biol Chem. 1998 May 29;273(22):13892-7. ↑
-
Cacabelos R. Eur Arch Psychiatry Clin Neurosci. 2008 Mar;258 Suppl 1:28-47. ↑
-
Polvikoski T, Sulkava R, Haltia M, et al. N Engl J Med. 1995 Nov 9;333(19):1242-7. ↑
-
Shah RS, Lee HG, et al. Biomed Pharmacother. 2008 March 17.[Epub ahead of print]. ↑
-
Shankar GM, Blood good BL, Townsend M, et al. J Neurosci. 2007 Mar 14;27(11):2866-75. ↑
-
Irvine GB, El-Agnaf OM, Shankar GM, et al. Mol Med. 2008 Mar 27 [Epub ahead of print]. ↑
-
Bouwman FH, Schoonenboom NS, et al. CSF biomarker levels in early and late onset Alzheimer’s disease. Neurobiology of Aging. 9 April 2008. ↑
-
Feng BY, Toyama BH, et al. Nat Chem Biol. 2008 Mar;4(3):197-9. ↑
-
Ghosh AK, Kumaragurubaran N, et al. Curr Alzheimer Res. 2008 Apr;5(2):121-31. ↑
-
Wolfe MS. Curr Alzheimer Res. 2008 Apr;5(2):158-64. ↑
-
Aisen PS, Gauthier S, et al. Curr Alzheimer Res. 2007 Sep;4(4):473-8. ↑
-
Santa-Maria I, Hernández F, et al. Mol Neurodegener. 2007 Sep 6;2:17. ↑
-
McShane R, Areosa Sastre A, Minakaran N. Memantine for dementia. Cochrane Database Syst Rev. 2006 Apr 19;(2):CD003154. ↑
-
Shah RS, Lee HG, et al. Biomed Pharmacother. 2008 Mar 17 [Epub ahead of print]. ↑
-
Cole GM, Teter B, Frautschy SA. Neuroprotective effects of curcumin. Adv Exp Med Biol. 2007;595:197-212. ↑
-
Begum A, Jones M, Lim G, et al. Curcumin Structure-Function, Bioavailability and Efficacy in Models of Neuroinflammation and Alzheimer’s. J Pharmacol Exp Ther. April 2008 Epub ahead of print]. ↑
-
Calabrese V, Butterfield DA, Stella AM. Nutritional antioxidants and the heme oxygenase pathway of stress tolerance: novel targets for neuroprotection in Alzheimer’s disease. Ital J Biochem. 2003 Dec;52(4):177-81. ↑
-
Goel A, Kunnumakkara A, Aggarwal B. Curcumin as “Curecumin”: From kitchen to clinic. Biochem Pharmacol. 75 (2008) 787-809. ↑
-
Mandel S, Amit T, Bar-Am O, et al. Iron dysregulation in Alzheimer’s disease: multimodal brain permeable iron chelating drugs, possessing neuroprotective-neurorescue and amyloid precursor protein-processing regulatory activities as the. Prog Neurobiol. 2007 Aug;82(6):348-60. ↑
-
agetia CG, Aggarwal BB. “Spicing up” of the immune system by curcumin. J Clin Immunol. 2007 Jan;27(1):19-35. ↑
-
Shishodia S, Singh T, Chaturvedi MM. Modulation of transcription factors by curcumin. Adv Exp Med Biol. 2007;595:127-48. ↑
-
Ono K, Hasegawa K, Naiki H, Yamada M. Curcumin has potent anti-amyloidogenic effects for Alzheimer’s beta-amyloid fibrils in vitro. J Neurosci Res. 2004 Mar 15;75(6):742-50. ↑
-
Yang F, Lim GP, Begum AN, et al. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005 Feb 18;280(7):5892-901. ↑
-
Garcia-Alloza M, Borrelli LA, Rozkalne A, et al. Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model. J Neurochem. 2007 Jul 27;102(4):1095-1104. ↑
-
Balasubramanian K. Molecular Orbital Basis for Yellow Curry Spice Curcumin’s Prevention of Alzheimer’s Disease. J. Agric. Food Chem. 54 (10), 3512 -3520, 2006. ↑
-
Cole GM, Teter B, Frautschy SA. Neuroprotective effects of curcumin. Adv Exp Med Biol. 2007;595:197-212. ↑
-
Cole GM, Morihara T, Lim GP, et al. Ann N Y Acad Sci. 2004 Dec;1035:68-84. ↑
-
Ganguli M, Chandra V, Kamboh MI, et al. Apolipoprotein E polymorphism and Alzheimer disease: The Indo-US Cross-National Dementia Study. Arch Neurol. 2000 Jun;57(6):824-30. ↑
-
Ng TP, Chiam PC, Lee T, et al. Curry consumption and cognitive function in the elderly. Am J Epidemiol. 2006;164:898–906. ↑
-
Fiala M, Liu PT, Espinosa-Jeffrey A, Rosenthal MJ, et al. Innate immunity and transcription of MGAT-III and Toll-like receptors in Alzheimer’s disease patients are improved by bisdemethoxycurcumin. Proc Natl Acad Sci U S A. 2007 Jul 31;104(31):12849-54. ↑
-
Fiala M, Cribbs DH, Rosenthal M, Bernard G. Phagocytosis of amyloid-beta and inflammation: two faces of innate immunity in Alzheimer’s disease. J Alzheimers Dis. 2007 Jul;11(4):457-63. ↑
-
Goel A, Kunnumakkara A, Aggarwal B. Curcumin as “Curecumin”: From kitchen to clinic. Biochem Pharmacol. 75 (2008) 787-809. ↑
-
Shoba G, Joy D, Joseph T, et al. Influence of piperine on the pharmacokinetics of curcumin in animals and human volunteers. Planta Med. 1998 May;64(4):353-6. ↑
-
Ono K, Yamada M. Antioxidant compounds have potent anti-fibrillogenic and fibril-destabilizing effects for alpha-synuclein fibrils in vitro. J Neurochem. 2006 Apr;97(1):105-15. ↑
-
Iuvone T, De Filippis D, Esposito G, et al. The spice sage and its active ingredient rosmarinic acid protect PC12 cells from amyloid-beta peptide-induced neurotoxicity. J Pharmacol Exp Ther. 2006 Jun;317(3):1143-9. ↑
-
Choi SH, Hur JM, Yang EJ, et al. Arch Pharm Res. 2008 Feb;31(2):183-7. ↑
-
Pereira P, Tysca D, Oliveira P, et al. Pharmacol Res. 2005 Sep;52(3):199-203. ↑
-
Yao Z, Deieu K, Papadopoulos V. The Ginkgo biloba extract EGb 761 rescues the PC12 neuronal cells from beta-amyloid-induced cell death by inhibiting the formation of beta-amyloid-derived diffusible neurotoxic ligands. Brain Res. 2001 Jan 19;889(1-2):181-90. ↑
-
Bate C, Salmona M, Williams A. J Neuroinflammation. 2004 May 11;1(1):4. ↑
-
Domoráková I, Burda J, Mechírová E, et al. Cell Mol Neurobiol. 2006 Oct-Nov;26(7-8):1193-204. ↑
-
Yao Z, Driew K, Papadopoulos V. The Ginkgo biloba extract EGb 761 rescues the PC12 neuronal cells from beta-amyloid-induced cell death by inhibiting the formation of beta-amyloid-derived diffusible neurotoxic ligands. Brain Res. 2001 Jan 19;889(1-2):181-90. ↑
-
Lyo Y, Smith JV, Paramasivam V, et al. Inhibition of amyloid-beta aggregation and caspase-3 activation by the Ginkgo biloba extract EGb761. Proc Natl Acad Sci U S A. 2002 Sep 17;99(19):12197-202. ↑
-
Yao Z, Han Z, Driew K, et al. Ginkgo biloba extract (Egb 761) inhibits β-amyloid production by lowering free cholesterol levels. J Nutr Biochem. 2004 15:749–756. ↑
-
Rudakewich M, Ba F, Benishin CG. Neurotrophic and neuroprotective actions of ginsenosides Rb(1) and Rg(1). Planta Med. 2001 Aug;67(6):533-7. ↑
-
Lee TF, Shiao YJ, Chen CF, et al. Effect of ginseng saponins on beta-amyloid-suppressed acetylcholine release from rat hippocampal slices. Planta Med. 2001 Oct;67(7):634-7. ↑
-
Li N, Liu B, Dluzen DE, Jin Y. Protective effects of ginsenoside Rg2 against glutamate-induced neurotoxicity in PC12 cells. J Ethnopharmacol. 2007 May 22;111(3):458-63. ↑
-
Joo SS, Lee DI. Arch Pharm Res. 2005 Oct;28(10):1164-9. ↑
-
Chen F, Eckman EA, Eckman CB. Reductions in levels of the Alzheimer’s amyloid beta peptide after oral administration of ginsenosides. FASEB J. 2006 Jun;20(8):1269-71. ↑
-
Lee TF, Shiao YJ, Chen CF, et al. Effect of ginseng saponins on beta-amyloid-suppressed acetylcholine release from rat hippocampal slices. Planta Med. 2001 Oct;67(7):634-7. ↑
-
Wang LC, Wang B, Ng SY, Lee TF. Effects of ginseng saponins on beta-amyloid-induced amnesia in rats. J Ethnopharmacol. 2006 Jan 3;103(1):103-8. ↑
-
Pereira C, Agostinho P, Moreira PI, et al. ↑
-
Wang R, Tang XC. Neurosignals. 2005;14(1-2):71-82. ↑
-
Little JT, Walsh S, Aisen PS. Expert Opin Investig Drugs. 2008 Feb;17(2):209-15. ↑
-
Zhang HY, Yan H, Tang XC. Non-cholinergic Effects of Huperzine A: Beyond Inhibition of Acetylcholinesterase. Cell Mol Neurobiol. 2008 Feb;28(2):173-83. ↑
-
Zhang HY, Tang XC. Neuroprotective effects of huperzine A: new therapeutic targets for neurodegenerative disease. Trends Pharmacol Sci. 2006 Dec;27(12):619-25. ↑
-
Ye JW, Cai JX, Wang LM, et al. Improving effects of huperzine A on spatial working memory in aged monkeys and young adult monkeys with experimental cognitive impairment. J Pharmacol Exp Ther. 1999 Feb;288(2):814-9. ↑
-
Sun QQ, Xu SS, Pan JL, et al. Huperzine-A capsules enhance memory and learning performance in 34 pairs of matched adolescent students. Zhongguo Yao Li Xue Bao. 1999 Jul;20(7):601-3. ↑
-
Shimmyo Y, Kihara T, Akaike A, et al. Three distinct neuroprotective functions of myricetin against glutamate-induced neuronal cell death: Involvement of direct inhibition of caspase-3. J Neurosci Res. 2008 Feb 8 [Epub ahead of print]. ↑
-
Shimmyo Y, Kihara T, Akaike A, et al. Multifunction of myricetin on A beta: neuroprotection via a conformational change of A beta and reduction of A beta via the interference of secretases. J Neurosci Res. 2008 Feb 1;86(2):368-77. ↑
-
http://www.phytochemicals.info/phytochemicals/curcumin.php ↑
-
Abdul HM, Butterfield DA. Protection against amyloid beta-peptide (1-42)-induced loss of phospholipid asymmetry in synaptosomal membranes by triclyodecan-9-xanthogenate (D609) and ferulic acid ethyl ester: Implications for Alz. Biochimica et Biohysica Acta. 2005 1741:140-148. ↑
-
Ono K, Horihata M, Yamada H. Ferulic acid destabilizes preformed β-amyloid fibrils in vitro. Biochem Biophys Res Com. 2005 336:444-449. ↑
-
Mamiya T, Kise M, Morikawa K. Ferulic acid attenuated cognitive deficits and increase in carbonyl proteins induced by buthionine-sulfoximine in mice. Neurosci Lettrs. 430 (2008) 115-118. ↑
-
Cho J, Sim H, Kim K, et al. Protection against β-amyloid peptide toxicity in vivo with long-term administration of ferulic acid. Br J Pharamcol. 2001 133,89-96. ↑
-
Kuyboyama T, Tohda C, Komatsu K. Neuritic regeneration and synaptic reconstruction induced by withanolide A. Br J Pharmacol. 2005 144:961-971. ↑
-
Tohda C, Kuboyama T, Komatsu K. Dendrite extension by methanol extract of Ashwagandha (roots of Withania somnifera) in SK-N-SH cells. Neuroreport. 2000 Jun 26;11(9):1981-5. ↑
-
Kuboyama T, Tohda C, Zhao J, Nakamura N, Hattori M, Komatsu K. Axon- or dendrite-predominant outgrowth induced by constituents from Ashwagandha. Neuroreport. 2002 Oct 7;13(14):1715-20. ↑
-
Kuboyama T, Tohda C, Komatsu K. Br J Pharmacol. 2005 Apr;144(7):961-71. ↑
-
Tohda C, Kuboyama T, Komatsu K. Neurosignals. 2005; 14(1-2):34-45. ↑
-
Kuboyama T, Tohda C, Komatsu K. Eur J Neurosci. 2006 Mar;23(6):1417-26. ↑
-
Schliebs R, Liebmann A, Bhattacharya SK, et al. Neurochem Int. 1997 Feb;30(2):181-90. ↑
{kind=link}
Comments are closed.